Cell therapy using embryonic or adult stem cells for regenerative medicine is generating high interest in the global medical community and in the general population.Physicians and patients are looking to cell therapies as potentially curative treatments for diseases such as diabetes, amyotrophic lateral sclerosis (ALS), Parkinson’s disease, Graft versus Host disease (GvHD), and cancer. Cell-based therapeutic products have been administered in clinics for nearly 90 years in the form of blood transfusions and for 50 years in the form of bone marrow transplants. With vast improvements over the past two decades in cell characterization, isolation, and in vitro manipulation, cell therapy has grown to include FDA-approved products such as
- cell-infused support matrices to regenerate damaged or injured skin
- investigative products for clinical trials such as cell-based vaccines for autoimmunity and cancer
- cell-based therapeutics for the treatment of cardiovascular, inflammatory, autoimmune, and neurodegenerative diseases and cancer. Furthermore, an increasing number of novel cell-based products for such indications have progressed closer to regulatory approval within the past three years.
PRODUCT FOCUS:CELL THERAPIES
PROCESS FOCUS:MANUFACTURING
WHO SHOULD READ: R&D, PROCESS DEVELOPMENT, QA/QC, MANUFACTURING
KEYWORDS: CELL ISOLATION, CELL PROCESSING, CLINICAL TRIALS, FACS (fluorescence-activated cell sorting), flow cytometery, CGMP
LEVEL:INTERMEDIATE
Cell therapy can be defined as the treatment or prevention of disease by administering cells that have been selected, manipulated, or altered outside the body. As more cell-based therapeutic products progress into clinical trials and commercialization, developing bioprocesses compliant with current good manufacturing practices (CGMP) has been challenging. This is because the final products are not traditional biological (secreted) molecules such as monoclonal antibodies but rather the cells themselves.
Industry focus has turned to one element of the cell-based product manufacturing process, cell isolation, because of the demonstrated importance of cell purity and the special considerations related to protocol compliance to CGMP regulations. Here, we compare technical aspects of fluorescence-activated cell sorting (FACS) and other cell isolation methods, summarize regulatory guidance for CGMP-compliant cell isolation, and provide points to consider when using FACS within a CGMP compliant manufacturing environment. This is not intended to be a comprehensive review of the field, but provides a basis for further reading and understanding.

General Steps in Cell-Based Product Manufacturing
The general steps for manufacturing a cell-based product are harvesting, debulking and isolation, ex vivo manipulation (e.g., activation, expansion, and/or genetic modification), and cryopreservation. Some cell-based products might be used after isolation without further manipulation or cryopreservation. Others might involve cell isolation after ex vivo manipulation. However, nearly all cell-based therapy products require some level of cell isolation or purification as part of the overall manufacturing process. These products are often sourced from heterogeneous cell populations with rare cells that need to be enriched or with contamination from unwanted cells that must be removed.
The production of regulatory T (Treg) cells, which are under study as a potential treatment for GvHD, offers an example of a selection strategy playing a key role in determining the activity and safety of a therapeutic product (1,2,3). Treg cells, which promote tolerance after allogeneic (donor) organ transplant or prevent GvHD after stem cell transplantation, share several cell surface antigens with alloreactive T cells, which are capable of causing GvHD in recipients. Isolation of Treg cells from peripheral blood using only a single antigen parameter results in contaminating alloreactive T cells. Furthermore, if the expansion of Treg cells is part of the manufacturing process, alloreactive T cells might be preferentially expanded under certain culture conditions, thereby resulting in lower therapeutic activity of the final product or unintended effects after patient administration (4,5,6).
Other examples in which cell purity is a factor include treatments based on human embryonic stem cells (hESC) and induced pluripotent stem (iPS) cell-derived products. Final products contaminated with undifferentiated hESC or iPS cells might form teratomas after injection (7,8). For some such issues, purification of specific therapeutic cell populations or removal of unwanted cell types during manufacturing might be critical in reducing some potential risks.
Cell Isolation and Processing Methods
Technical Considerations: Selecting elements of an overall cell isolation process depends on the relative abundance of the cells of interest in the source material and the number of cells and level of purity needed for an intermediate or final product. The best available and/or most practical method to separate cells of interest from unwanted cells must also be taken into account. There are three general approaches to cell isolation: Cells can be separated on the basis of their physical properties (e.g., density, size), biological or genetic properties (e.g., adherence to plastic, drug resistance), or cell-surface antigen expression (immunophenotype). Figure 1 shows a typical decision tree isolating several cell types from blood or bone marrow. Each manufacturer of therapeutic cells must develop, optimize, and validate its own process based on specific tissue source, cell type, and final product needs.
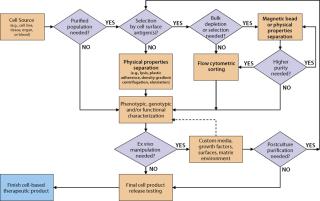
For solid tissue sources, a dissociation step using mechanical and/or enzymatic technologies may be performed before debulking the cells. The source and quality of enzyme reagents is an important consideration for processes leading to clinical products. Enzyme reagents derived from animal sources should be tested for potential adventitious agents. For bone marrow, peripheral and cord blood, and other liquid or semiliquid sources, debulking might be performed after harvest by physical property approaches such as density gradient centrifugation or elutriation. Separation methods based on physical properties generally have the capacity to process or debulk very large numbers of cells in a single step (e.g., 1–3 × 1010 input cells). However, they lack the ability to precisely isolate or deplete specific subpopulations of a given cell type and often result in a heterogeneous mix of cells at the end of the process (Table 1). Some clinical cell processes already involve several commercial systems, and developers need to consider instrument performance specifications to select the best method for their needs.
Table 1: Comparison of cell isolation approaches
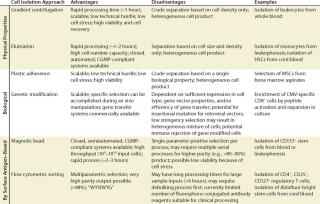
Cell separation using biological properties is also amenable to processing large numbers of input cells. A common method is differential expansion of a specific cell subtype in culture. Selective conditions that can be optimized for desired cell types include specific growth factors, cytokines and substrates, length of culture, support matrix, temperature, and/or feeder cell lines. For naturally adherent cell types such as mesenchymal stem cells (MSC), one approach to debulking and isolation is culturing them on an appropriate solid surface and washing off nonadherent cells. Biological selection also can be used on genetically modified cells.
One approach to achieving selection, involves inclusion of a drug-resistance gene that imparts specific resistance to an antibiotic or other compound. Addition of such a drug to cell cultures would inhibit the survival of nonmodified cells, resulting in the enrichment of the desired cells. Alternatively, you could achieve positive selection of cells modified with a receptor gene if a ligand, antigen, or tetramer specifically expands the gene modified cells in culture. Several serum-free, commercial media are highly defined and already used in many CGMP-compliant bioprocesses. Media supplements, cytokines and growth factors, and feeder cell lines should all be tested and qualified for cell-based product manufacturing similar to what is performed for other clinical biological products. One advantage of biological property–based selection is that it can be performed during ex vivo manipulations such as activation or expansion. However, like separations based on physical properties, these approaches often yield a mixture of cells, and the level of contaminating cells depends on the stringency of the selection system.
For cell types with known cell surface antigen phenotypes, more directed cell isolation technology is based on specific antigen-antibody binding interactions. Antibody-coated magnetic bead reagents can be used to positively select or deplete cells displaying a specific surface antigen. This approach can process a high number of cells in a few hours, but the resulting product output might still be a heterogeneous mixture of cells because of the single parameter selection. For cell-based products requiring a higher level of purity, a series of magnetic bead–based separations or a single separation with a bead cocktail containing several specificities can remove unwanted cell types. A secondary positive selection after depletion might provide further purification. However, that approach might not be practical for highly defined cell populations because of the overall process yield, the availability of antibody specificities of the bead reagents, and the cost of reagents.
Because FACS analyzes and isolates cells individually on the basis of multiple parameters for each cell, it can achieve levels of purity of the cells of interest that are often not possible through magnetic bead–based separations (Figure 2). A single process can remove unwanted cells and isolate specific populations based on a complex cell surface phenotype. FACS also can characterize the exact population of cells isolated for quality control (QC) purposes. Nevertheless, this approach might be limited by a flow cytometer’s sort rate, which can make isolating a rare cell subpopulation from a very large starting sample impractical. But for cells that are abundant in the starting material or processes needing 10–100 million processed cells, FACS generates highly defined, purified cell preparations with a low number of unwanted cells in the final product.

In addition to purity, yield, and throughput, cell-product manufacturers are concerned about postisolation viability, which has a significant impact on downstream cell-manipulation processes and final product quality. All the other isolation methods described thus far have some influence on cell viability and function. Density-gradient centrifugation and elutriation are gentle methods that yield a high percent of viable cells. The postisolation viability of cells isolated using magnetic bead systems or FACS depends on a number of factors, including temperature, pH, aggregate formation, medium formulation, excessive cell handling, adherence, and shear forces.
CGMP Compliance: Cell isolation methods require not only consideration of their technical aspects, but also their amenability or adaptability to being CGMP-compliant processes. The meaning of CGMP often causes confusion in industry because it is commonly misused to mean that a product manufactured according to CGMP is “clinical grade” and appropriate to administer to patients. This is incorrect because Chapter 21 of the United States Code of Federal Regulations (CFR), the source of CGMP regulations, applies them to the manufacturing of products ranging from drugs for human use to medicated animal feed. Rather, CGMP should be viewed as the current minimum requirements for the methods, facilities, and controls used in manufacturing, processing, and packaging of drugs and other FDA-regulated products to ensure their safety and quality.
21 CFR parts 210, 211, and 600-610 describe the regulations for CGMP drugs, including cell-based products. These sections discuss the manufacturing, processing, and holding of drugs; finished drugs; and blood and biological products, respectively. In addition to CGMP regulations, 21 CFR includes pertinent regulations for cell-based products, including part 1271 for human cells, tissues, and cellular and tissue-based products. US facilities undertaking the manufacture of cell-based products might want to consult the Federal Register for new and updated CGMP regulations and other documents such as Guidance for Human Somatic Cell and Gene Therapy (March 1998), as well as the regulations and guidances promulgated by the Center for Biologic Evaluation and Research (CBER) to manufacture and process vaccines, blood, and biologics. Furthermore, they should review US Pharmacopeia (USP) standards for ancillary materials used to manufacture cell products. Finally, bioprocess engineers should review local (state or county) regulations that might also apply (e.g., local pharmacy laws and regulations).
Outside the United States, other regulatory requirements might apply in addition to (or in lieu of) FDA regulations. In Europe, medicinal products are evaluated by the European Medicines Agency (EMEA). The Committee for Advanced Therapies (CAT) provides specific guidance for cell-based products in Regulation (EC) No. 1394/2007 on advanced therapy medicinal products. Countries might have their own regulatory agencies or may adopt regulatory requirements from the United States or Europe. The International Standards Organization (ISO), which strives to set worldwide standards by facilitating harmonization of regulations from different countries, might have applicable guidance.
Interested readers should consult with regulatory professionals specialized in these areas of expertise before embarking on cell processing for therapeutic purposes.
Manufacturing a Cell-Based Product
When designing a manufacturing process for a cell-based product, developers must consider the performance specifications of the isolation and purification technology that best meets their product and process needs. The ability of each technology to comply with CGMP regulations should be carefully reviewed. Researchers can perform both elutriation and magnetic bead–based approaches using commercial instruments with closed, single-use systems (e.g., CaridianBCT Elutra separation system and Miltenyi CliniMACS Plus instrument, respectively) and clinical-grade reagents (e.g., Miltenyi CliniMACS magnetic beads), which have already been used to isolate several cell-based investigative products. Most commercial FACS instruments do not have closed, single-use fluidics systems. However, recently introduced instruments (e.g., BD Influx) have fluidics that are easily exchangeable. Replacement of a fluidic system from sort nozzle to a sheath reservoir between samples is intended to facilitate compliance with CGMP regulations. Ultimately, manufacturers should take into account available technology, their desired outcomes, and the practicality within CGMP regulations for a specific process.
FACS for Cell Processing: FACS has been used extensively in basic and preclinical research to isolate specific cell populations of interest so their potential as a therapy could be investigated and evaluated. When used to isolate therapeutic cells in clinical trials or in commercial settings, FACS must adhere to the relevant aforementioned regulations and general guidelines for cell processing. Table 2 lists key issues and regulations that cell processing engineers and manufacturers should take into account, including sample contamination and line clearance, cytometer setup, operator protection, reagent quality, and final product QC (Table 2).
Table 2: Example of some regulatory considerations for FACS of cell-based products
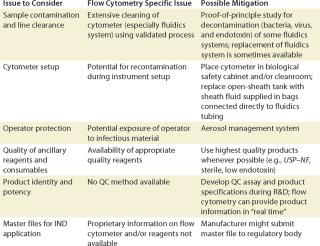
Sample Contamination: The cross contamination of a FACS-isolated cell-based product by a preceding processed product is a major concern to regulatory agencies because such cross contamination can inadvertently transmit disease or taint a final product. To minimize cross-contamination risks, line clearance (as defined by CGMP) is required. Line clearance of a flow cytometer removes all traces of previous sample, microbial and/or endotoxin contamination, and all traces of harmful cleaning materials. Such line clearance and validation of a process can be extremely difficult for flow cytometers, considering that the fluidics system might contain small areas where contaminating cells and microbes can escape removal or inactivation by the decontamination procedure.
Proof-of-principle research studies have been undertaken to demonstrate the effectiveness of a specific prepare-for-aseptic-sort (PAS) procedure. These research studies show that a bacteria-free, low endotoxin fluid path might be achieved in certain FACS instruments (8,9,10).
Another approach involves complete replacement of the fluidics path that comes in contact with a biological sample, thus eliminating the need for line clearance and development of validated cleaning procedures. Recently introduced instruments (e.g., the BD Influx cell sorter) have fluidic systems from sort nozzle to sheath reservoir that are easily exchangeable.
Cytometer Set-Up: Setting up a cytometer before cell sorting can inadvertently reintroduce contamination even if the fluidic system has been previously cleaned or replaced.
For a FACS instrument fitted with an open sheath tank, operators should clean and sterilize the tank using validated procedures and refill it with appropriate quality sheath fluid using aseptic techniques. Tubing, connectors, and filters should be cleaned or replaced, and the cytometer should be aseptically connected. In addition, all instrument surfaces that might expose a cell product to microbial or other contamination should be cleaned. Pressurization of the sheath fluid by a pump or pressurized air system can introduce contamination unless the fluid is protected by an appropriate quality air filtration system.
One potential solution to that risk is the sterile connection of sheath fluid supplied in bags using a tube connection device. A bag of sheath fluid can be hung inside a sheath tank that is pressurized during operation, which enables a fluid to flow throughout the path without direct contact with the pressurization system.
Operator Protection: Sorting biological specimens using a FACS instrument presents unique challenges to protect an operator from potentially infectious materials that might be contained within cell-based raw material. These materials may harbor infectious agents such as viruses, viral or bacterial gene vectors, bacteria, fungi, and prions. Cell sorting generates droplets and aerosols that might be inhaled by an operator and cause infection by means of mucous membranes in the respiratory pathway. For this reason, the International Society for Analytical Cytology (ISAC) has developed a safety standard for sorting unfixed cells (11).
The risk of exposure to potentially infectious material should always be taken into account during development of a sorting procedure for isolating cell-based products. One of the most effective methods for containment is an aerosol management system that removes droplets and aerosols generated during sorting. For example, the entire instrument or components that potentially generate aerosols might be placed inside a Class 100 biosafety cabinet. In all cases, the efficacy of an aerosol management system must be validated in situ. The ISAC biosafety standard provides guidelines on validating an aerosol management system.
Reagent and Other Ancillary Product Quality: The quality of reagents used during a sorting procedure should be chosen carefully. They need to be appropriate for the product being manufactured as specified by local, regional, and national regulatory agencies. For example, although research-use-only (RUO) reagents might be of sufficient quality to sort cells for in vitro research, higher quality reagents are recommended for cells sorted for in vivo research studies, especially if research data might be used to design clinical studies.
Furthermore, manufacture of clinical cell-based therapy products should involve only the highest quality reagents, in accordance with regulatory guidances. For example, fluorophore-conjugated antibodies for performing FACS can be produced from serum-free adapted cells and tested for sterility, toxicity, and the presence of adventitious agents because they directly contact a cell-based product. Even ancillary and consumable products that do not directly contact a cell-based product should be of appropriate quality. Ancillary and consumable products include antibodies used to label the cells before sorting, plastic components of a cell sorter’s fluidics system, and buffers used to wash the cells. The USP provides standards for materials involved in manufacturing cell-based products, and manufacturers should refer to their local or national regulatory agencies for additional guidance.
Quality Control of a cell manufacturing process is central to produce a high quality cell-based product and should be designed with careful reference to regulations such as CGMP. Product QC might require evaluating research-based procedures during product development, which can be the groundwork for designing product QC assays, setting specifications and, ultimately, a monograph in USP or equivalent. Several monographs for biotechnological or biological products are already available, and USP contains guidelines on various aspects of the QC for these products.
One key aspect of cell-based product QC is complete characterization of the product, not only with respect to the identity and purity of the primary cells, but also with respect to other cells that might contaminate it. One advantage of an FACS instrument is that its cytometer software can track cell product characteristics (e.g., identity, purity, and yields) in real time. That information can provide in-process QC of the product during a portion of a manufacturing process. All other methods of cell isolation require product sampling and performing a separate assay to characterize intermediate and final products.
Master Files and Investigational New Drug Applications: For a US market, a FACS instrument, consumables, and ancillary reagents should be well described, particularly in the chemistry, manufacturing, and controls (CMC) section of investigational new drug (IND) applications. Detailed information that may be requested by a regulatory agency or internal review board might be confidential or unavailable to the public. To facilitate use of their instruments and reagents in qualified clinical studies, some manufacturers submit product master files to FDA or equivalent regulatory body. These files contain proprietary information about a product’s manufacturing process, reagents and components, testing, and specifications. Clinical investigators can reference this information in their INDs, thus reducing investigator’s burden of paperwork. When developing a clinical manufacturing protocol, investigators might want to contact FACS instrument and reagent manufacturers to determine whether master files have been submitted for instruments and reagents of interest.
FACS AND FLOW CYTOMETERY
Fluorescence-activated cell sorting (FACS) is a specialized type of flow cytometry. The acronym is trademarked by Becton Dickinson. Although many people use it for all types of cell sorting and related applications, it is not a generic term for flow cytometry.
The first cell sorter was invented by Mack Fulwyler in 1965, who used a relatively difficult technique not applicable to modern instruments. His technique was expanded by Len Herzenberg, who coined the term FACS and won the 2006 Kyoto Prize for his work in flow cytometry. The type of fluorescent label used depends on available detectors as well as the lamp or laser used to excite fluorochromes.
Other Flow Cytometers
The Guava easyCyte HT family from Millipore Corporation (www.millipore.com) includes patented microcapillary technology that operates without sheath fluid and allows for extremely small sample and waste volumes.
Gallios flow cytometers from Beckman Coulter (www.coulterflow.com) are intended for research use only (not for diagnostic procedures). Coulter Epics XL and XL-MCL flow cytometers are intended for high-throughput clinical applications. And Beckman Coulter’s MoFlo XDP cell sorter is intended to compete directly with BD FACS Aria systems.
The Sony company iCyt (www.i-cyt.com) offers the Reflection parallel cell sorter and the Eclipse cell analyzer and counter. And Accuri Cytometers Inc. (www.accuricytometers.com) offers the C6 flow cytometer system.
—Cheryl Scott, senior technical editor
Flow Cytometric Sorting for Cell Therapy: The potential of novel cell-based therapeutic products to treat or cure a multitude of diseases that have currently poor or no treatment options has excited the medical and general communities. Manufacturing processes are needed to efficiently and safely produce cell-based therapeutic products for clinical trial evaluation and eventually commercial use. We highlighted here several technical and regulatory issues that cell-based product developers should consider if cell isolation is part of their manufacturing processes. For complex cell products that require high purity and defined populations, FACS is an isolation method that can multiparametrically select individual cells and record the process of doing so in real-time. Flow cytometers can be used in clinical drug manufacturing only if instruments, reagents, and overall processes comply with CGMP and regulatory guidance from local and national agencies. Although no single instrument is currently specifically designed to meet all aspects of clinical manufacturing guidelines and principles, several commercial cytometers can be and have been adapted for this use. Design improvements such as exchangeable fluidics path and biosafety cabinet enclosures on newer models have facilitated application of cytometers in a CGMP environment. Such instruments, combined with the availability of ancillary reagents such as fluorochrome conjugated antibodies, manufactured according to CGMP, have enabled clinical investigators to isolate highly defined cell populations for clinical use, bringing cell therapies closer to reality.
Disclaimer: We make no claims that the list of regulations and guidances listed in this document is comprehensive but merely offer an overview of the complexity of the laws and regulations that might pertain to cell processing for therapeutic purposes. We highly recommend that interested readers consult with regulatory professionals specialized in these areas of expertise before embarking on cell processing with the intent of using processed cells for therapeutic purposes.
Author Details
Catherine A. McIntyre, PhD, is manager, systems validation and application notes, at BD Biosciences; Brian T. Flyg, MS, MBA is strategic marketing manager at BD; and corresponding author Timothy C. Fong, PhD, MBA, is technical director of the cell therapy research program at BD Biosciences, 2350 Qume Drive, MC B2B-018, San Jose, CA 95131; 1-408-954-2528, fax 1-408-954-4122; tim_fong@bd.com; www.bd.com.
REFERENCES