Challenges and approaches in demonstrating comparability of a well-characterized biotechnology product after manufacturing changes can be as varied and complex as the products themselves. Participants at the January 2005 CMC Strategy Forum sought to discuss and agree on common implementation strategies for different manufacturing change scenarios. Development of flexible, comprehensive approaches in strategy development addressed evaluation of critical product characteristics, appropriate process steps to test, numbers of lots and levels of testing required, and assessment of product comparability (e.g., trending analysis, additional characterization studies, accelerated stability data). The change scenarios we discussed can occur throughout the life-cycle of a product from early development through postapproval manufacturing.
PRODUCT FOCUS: WELL-CHARACTERIZED BIOLOGICS
PROCESS FOCUS: UPSTREAM AND DOWNSTREAM PROCESS DEVELOPMENT
WHO SHOULD READ: MANUFACTURING, PROCESS DEVELOPMENT, ANALYTICAL, AND PRODUCT DEVELOPMENT PERSONNEL
KEYWORDS: CHARACTERIZATION, CHANGE CONTROL, SPECIFICATIONS, DESIGN SPACE
LEVEL: INTERMEDIATE
Early stage development is where the foundation for assessing comparability begins, and the effects of good or poor development will carry throughout a product life-cycle. Sufficient process and product knowledge is required for reliably predicting and assessing the impact of a change and ensuring that a product will consistently meet approved specifications and standards. Efforts required to assess comparability are inversely proportional to a manufacturer’s understanding of its manufacturing process, product quality attributes, and capability of the analytical methods used. An assessment of comparability should show that products are highly similar before and after a manufacturing change occurred. It does not mean the products are identical, but that their physicochemical and biological properties are sufficiently similar to ensure no adverse impact on their quality, safety, or efficacy.
Demonstrating Comparability
Forum Attendees Unanimously Agree: “Change Is Good!” The intention of this forum was to clarify for manufacturers the appropriate factors to consider when making changes to operations in pre- and postapproved manufacturing processes while ensuring patient safety, drug efficacy, and product quality. With all the issues to consider in demonstrating comparability, attendees unanimously agreed that despite associated hassles, the benefits of making changes far outweigh the costs of comparability studies.

WWW.PHOTOS.COM
This familiar conundrum is reminiscent of a quote by famed cartoonist Sydney Harris: “Our dilemma is that we hate change and love it at the same time; what we want is for things to remain the same but get better.” As process knowledge and new technologies improve over time, manufacturers must follow. The needs of patients and caregivers also change over time and might require altering formulations or product presentations.
Regulatory agencies have agreed that it is reasonable for manufacturers to make changes in the steps of their manufacturing processes or specifications (testing and/or acceptance criteria) over the life-cycle of a biopharmaceutical product. Of the many benefits discussed at the forum, the main reasons for introducing process changes included process economics, product quality improvements, manufacturing technology advances, production yield or overall global capacity increases, and global harmonization of operating parameters. Improvements in test methods for process control and product characterization have driven evolution of the regulations. Innovation and improvements in manufacturing processes and test methods have been encouraged because they bring important and improved products to market efficiently and expeditiously. It was agreed that change is necessary for the biotech industry to evolve and progress, and international regulatory agencies see that as very positive outcome.
For well-characterized biotechnology products, comparability is the demonstration or existence of a high degree of similarity between products made by different manufacturing processes. Changes should have no adverse effects on the quality, safety, or efficacy of a product — the ultimate bottom line for demonstrating comparability. Several existing FDA, EU, and ICH guidance documents describe the principles of demonstrating comparability (1,2,3,4). A central tenet of those principles is that a given manufacturing change can be adequately assessed by comparing pre- and postchange materials and demonstrating that they are comparable (postchange material is of the same or better quality).
One forum attendee noted that for biologically derived products, better quality does not always mean greater purity. In certain products, impurities can act as stabilizers or enhance or inhibit activity. For example, attendees cited how a purer product may aggregate or cause an immunogenic response (as can a less-pure product).
Regulatory agencies encourage manufacturers to perform relatively extensive characterization in early phase development. The rationale is that such studies will be the primary focus in physicochemical characterization because not much information is known about clinical development and trials at that point. A thorough understanding of how manufacturing changes affect product quality attributes is desirable early in development because product–process relationships are important throughout the life-cycle of every product. Attendees acknowledged that obtaining early characterization data is resource dependent but often advantageous to manufacturers. Even if one product does not succeed to market, often a company can apply learning from it to similar modalities. (Monoclonal antibodies were cited as example.)
Changes in product or process may be more acceptable at early stages than after phase 3. Michael Klein of Amgen provided an excellent example of comparability assessment for a manufacturing change in early product development. The FDA’s decision on comparability for the example was that the new product was sufficiently similar (though not comparable) to that used in earlier studies for its use to be allowed. Because the altered product was subject to further clinical evaluation, the potential impact of changes on safety and efficacy would be addressed. At later phases of development and postapproval, more stringent comparability criteria may be warranted.
PROCEEDINGS OF THE WCBP CMC STRATEGY FORUM, 9 JANUARY 2005
The fifth Well-Characterized Biotechnology Products (WCBP) Chemistry, Manufacturing, and Controls (CMC) Strategy Forum was held on 9 January 2005 at the Renaissance Mayflower Hotel in Washington, DC. Its purpose was discussion of issues related to demonstrating comparability for well-characterized biotechnology products in early and late development phases and postapproval. As with previous CMC Strategy Forums, the California Separation Sciences Society (CASSS; www.casss.org) sponsored this event. The organizers and moderators were John Towns (Eli Lilly) and Keith Webber (CDER, FDA). More than 130 attendees represented 40 large and small companies and 15 consulting firms as well as government agencies and academic organizations.
The forum consisted of a morning session devoted to the demonstration of comparability during preapproval and an afternoon session focused on postapproval comparability. Webber pr
ovided opening comments regarding the FDA’s current regulatory guidelines and perspective related to demonstration of comparability. Michael Klein of Amgen provided an example of the evaluation of comparability preapproval by pointing to elements crucial to successful design and implementation of demonstrating comparability for product changes preapproval. Attendees then received three questions related to preapproval comparability that facilitated discussion for the remainder of the session.
The afternoon session began with two case studies by Tina Norsell of Novo Nordisk on the investigation of impurities in a biopharmaceutical product. Genentech’s John O’Conner followed with a presentation on demonstrating comparability for products manufactured at different global facilities. Allison Wolf of Eli Lilly provided a case study for successfully reducing a product’s reporting category by using a comparability protocol to reduce the reporting category down from a prior-approval supplement (PAS) to an annual reportable (AR) change. Attendees then received three questions relating to postapproval comparability that facilitated discussion for the remainder of this session.
Bioassays and/or pharmacokinetic and pharmacodynamic studies may be required for biotechnology-derived products because many physicochemical tests cannot accurately detect small changes in such products. Animal testing may be required if in vitro bioassays do not detect potential changes in a product’s tertiary structure. If physicochemical comparability cannot be demonstrated with a production batch, then the change has substantial potential to adversely affect the identity, strength, quality, purity, and/or potency of a drug product. An applicant still wishing to institute such a change would perform additional studies to assess the impact of that change. Those might include in vitro or in vivo biological studies or even human clinical trials.
Attendees emphasized that postchange statistical impurity limits should be within the range experienced in clinical trials. When impurities reach levels beyond what was observed clinically, the decision most likely to be made is to alter a process further and get those impurities back within the range used for clinical trials. Also, with increased manufacturing experience, the variance of product characteristics often decreases. Manufacturers may consider narrowed ranges as a benchmark during comparability assessment if that helps them ensure consistent product quality.
Implementing substantial manufacturing process changes after phase 3 is considered a special case that generally requires submission of data to demonstrate comparability in a marketing application. The FDA cited such changes as a special case because they come after completion of product safety and efficacy studies. The agency realizes that it is not always possible to delay implementing a change, but doing so increases the amount of information that must be reviewed prior to approval.
Attendees said that major failures in demonstrating comparability for a change were often caused by inadequate training of new people as well as subtle differences in new equipment and raw materials. Under the training of personnel, a subtopic of transferring analytical methods was cited as a large cause of variability when making a manufacturing site change. In extreme cases methods could not be transferred, so originating laboratories have been stuck running a new site’s tests. Even hiccups extrinsic to a process (e.g., a faulty flow valve with a column resin change) need to be investigated and resolved when the flow of consecutive lots is interrupted. Another example cited occurred when the composition of media needed to be adjusted during demonstration lots to obtain a similar glycosylation pattern (in this case galactose levels) to that observed before a manufacturing change. Lower galactose levels were not a problem of the expression system but came from stress the cells were under as the manufacturer worked toward higher titers. Higher galactose levels were easily achieved by increasing certain nutrient levels in the media.
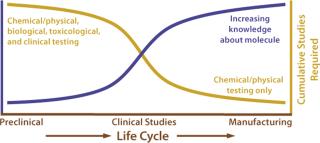
Figure 1 shows the knowledge base and testing curves as a function of a product life-cycle. The knowledge base increases as a manufacturer learns more about critical process parameters and product quality attributes. Meanwhile, overall testing (physicochemical, biological, toxicological, and clinical) decreases. Early development involves a full arsenal of testing from animal tests to clinical trials and physicochemical and/or biological testing. As the knowledge base increases, the testing curve decreases. In assessments of comparability for postapproval manufacturing changes, the testing requirements often can be limited to physical, chemical, and biological testing.
Risk-Based Approach: Any modification to the method of manufacture for a drug substance carries some risk of causing adverse effects either to the physical properties of the molecule or the level and nature of impurities present (5, 6). The molecular complexity of biopharmaceuticals presents unique challenges in assessing the impact of a given manufacturing change. Relatively small changes can alter the characteristics of a drug substance and thus affect safety and efficacy (7,8,9). Of particular concern is the potential for manufacturing changes that increase product immunogenicity. Companies also should consider the potential impact that a change at one point in a manufacturing scheme can have on other steps in the same process.
An FDA representative stressed a risk-based approach in the evaluation of comparability and encouraged industry to do so as well. In thinking about making a process change, a manufacturer should identify potential alterations of impurities or the product itself. Companies will use their knowledge of process and product to assess the risk that changes may negatively affect quality, safety, or efficacy of the product. Based on such risk assessment, a manufacturer can evaluate how extensive its studies should be in determining comparability. For example, a major change in culture media composition could have significant impact on the protein product as well as its impurities — so related comparability studies should be comprehensive. On the other hand, changing a chromatography column packing from one brand to another may not require such extensive comparability assessment.
On the subject of “how we can really make all our lives easier,” one attendee said that the best way to handle process changes is to eliminate the need to make them in the first place. By conducting process development studies in sufficient depth, a manufacturer might not have to make many changes in the future. For example, it could start with a commercially viable cell line at phase 1. Attendees agreed that doing so requires a certain amount of investment, but in the long run it is probably a better approach than dealing with comparability assessment for a plethora of changes just before and immediately after commercialization. Questions from the Preapproval Session
Question 1: What factors (e.g., safety, physicochemical attributes) need to be considered to demonstrate comparability at different phases of clinical development? Seven factors rose to the foref
ront of this discussion:
Extent of change: Extensive changes to a process or those made to critical steps (e.g., cell line or cell source) must be considered.
Phase of development: Early phase changes have less potential for concern because a drug is still being tested.
Mechanism of action: Changes in critical steps of production that might affect the molecule’s function (e.g., glycosylation) must be considered.
Ability to assess change: The manufacturer’s ability to appropriately assess the impact of a change needs to be considered. New tests may need to be developed.
Downstream processing and clean-up: The ability of downstream process steps to remove process-related and/or product-related impurities is important.
Knowledge base: Manufacturers must understand critical process parameters and product quality attributes to focus their comparability studies.
Patient population factors into the risk-benefit assessment. For example, immunocompromised patients may be less likely to develop an immune response to protein therapeutic products.
Question 2: What comparability testing requirements are necessary for dosage-form and formulation changes during clinical development? One main challenge in product development is setting meaningful acceptance criteria for product attributes with which a manufacturer has limited experience. It can be difficult to ensure that material bridging to a new formulation or dosage form is actually comparable to what was used before.
One attendee presented a case study regarding a change from a liquid formulation to a lyophilized product. Susceptibility of a protein product to aggregation from interactions with excipients in a new formulation can have a large impact on product stability or bioavailability. Effects of excipients on freeze–thaw and product storage were also cited as benefiting from comparability testing. Discussed in detail was the use of surfactants such as Tween in protein formulations. Although it is used to prevent aggregation, some cited Tween as causing the opposite effect above a critical micelle concentration. It was also cited as a raw material that contained potential contaminants, such as higher levels of residual peroxide, which could adversely affect product stability. Attendees were advised that if changes involved such excipients, it might be best to consider additional raw-material testing beyond that usually provided by suppliers. One highly publicized example is a formulation change involving Tween that may have affected product quality and caused the development of antibodies against epoetin (10).
Attendees also cited container–closure interactions as a problem. An example was the move from a solution formulation in a vial to a prefilled syringe. The potential for leachates can be a concern, whether from a vial, stopper, or a syringe. In addition to rubber and metal leachables, problems can come from gluing components found in stake-needle syringes. Such contact surfaces have led to protein modifications and thus could require development of new analytical testing.
Tina Norsell of Novo Nordisk provided two case studies of investigations centered on identifying a contaminant, locating the root cause of it, and determining patient safety. She spoke of contaminants extracted from tubing used in a manufacturing process and from rubber stoppers used during freeze drying. In the former study, the contaminant was first tracked down to wastewater from a washing machine after equipment cleaning. That impurity was identified as 2,4-dichlorobenzoic acid (DCBA) and was only 0.07% of the total peak area in a drug product purity chromatogram. Toxicological and health-hazard evaluations concluded that it was not associated with any risk to human health. Silicone tubing made of peroxide-cured silicone can leach out DCBA, whereas platinum-cured silicone does not. The investigation was difficult because of higher leachate levels in the first solutions to go through the tubing. They greatly decreased as more solution was sent through.
In the second Novo Nordisk study, all components of a freeze-dryer — gaskets, vacuum oil, and so on — were analyzed along with an extraction of the primary packaging material. During the freeze-drying process, xylene was coming from the rubber stoppers. Xylene is listed in an ICH guideline with a concentration limit of 2,170 ppm (6). The amounts found were much lower than that limit, so it was determined not to be a patient-safety issue. These examples illustrate that trace amounts of chemical contaminants do occur and that analytical testing capabilities are continuously improving to detect lower and lower impurity concentrations.
Question 3: How do I establish critical product characteristics in demonstrating comparability? Forum attendees agreed that a detailed comparability plan listing the acceptance criteria of critical process parameters and key product quality attributes is critical to successfully executing a manufacturing change. A comparability plan flows out of this understanding and is a key discussion item with the regulatory agencies. As a rule, manufacturers should provide adequate chemical and physical (and in some cases biological) comparisons, with side-by-side analyses of the “old” and “new” materials and demonstrating that postchange material is comparable to prechange material. The array of product attributes evaluated within the comparability plan will depend on knowledge of product structure–function relationships. An FDA representative at the forum stated that the agency does take into consideration what information manufacturers have at small scales as well as development data in evaluating a total comparability package to justify the lack of adverse affects from a change. Testing Strategy and Criteria
Critical product-characteristic requirements for demonstrating comparability have focused both on testing strategies (what to test, where to test, and how many lots) and on the setting of meaningful acceptance criteria.
What to Test: In determining the types of tests needed, consider the extent of your manufacturing change(s) and the stage of manufacturing at which it occurs. Knowledge and understanding of the manufacturing process are integral components in determining the design of an appropriate assessment program. Ensuring conformance with final-product specifications is the primary concern in demonstrating comparability; however, conformance with drug-substance specifications or in-process control criteria may also be critical for upstream changes (11, 12). Of special consideration are tests contained within the pharmacopeial monographs. Some factors involved with a given product may be unique, and testing might be required beyond those listed as compendia criteria.
Characterization tests beyond those used for lot release are important for demonstrating comparability and should include expanded biochemical characterization. Consider product-related substances and impurities (e.g., deamidation, oxidation, and glycosylation) in addition to process-related impurities (e.g., host-cell proteins, DNA, and residual solvents).
Establishment of reference standards as early as possible is critical to the success of comparability testing. It is difficult to demonstrate comparability without standards, in which case a manufacturer lacks a starting point to compare products before and after a change. Retention samples from production batches also can be valuable for establishing reference points within the current manufacturing process.
Where to Focus an Evaluation: In addition to evaluating effects at the process step being changed, consider also the potential for effects on subsequent steps, including drug substance and drug product. Ideally, impurities should be ev
aluated in intermediate materials immediately following the process step in which a manufacturing modification was made. A process step further downstream may be selected if a historical database of process-related impurities exists for that step.
How Many Lots: Historically, three lots of postchange material have been compared with three or more lots of prechange material. The FDA looks at the type of change to determine how much justification is needed in making it based on the level of information on hand and a demonstrated lack of adverse impact with a minimum number of lots. In some cases, one lot has been acceptable for minor changes. The amount of process- and product-development data is a factor in determining how much additional information is needed to support a process change. Non-GMP lot history may be helpful in establishing a reduced number of GMP lots to demonstrate comparability.
Additional testing (beyond what is used for lot release) is often needed to assess whether or not material differences exist before and after a change. One attendee reported having supported a recent manufacturing change by comparative testing with three lots of new API and all the previous year’s campaign (30 lots) of historical limits based on 3-sigma and product-related substance testing results that showed no new impurities.
Design Space and Comparability Protocols: One approach proposed by an FDA representative is defining an appropriate “design space,” within which a company can vary its manufacturing process without its being considered a “change” by the regulators. Alternatively, a protocol can describe how the effects of manufacturing changes will be evaluated, including related acceptance criteria. That can allow multiple changes to be implemented over time under a single comparability plan. Under the new paradigm, specifications focus on defining essential product quality attributes that relate to safety and efficacy of a drug product, whereas process controls serve to ensure that the product conforms with its specifications. Questions from the Postapproval Session
Question 1: What amount (conditions and length) of stability testing is appropriate? Assessment of pre- and post-change comparability is likely to benefit from and include the need for stability testing of a drug substance and/or drug product (13,14,15). Stability testing is a cornerstone of comparability demonstration. It provides an assessment of both the degradation of materials and the likelihood that a drug substance or product will stay within its end-of-shelf-life specifications. Assessment of stability is likely to involve studies under two conditions: accelerated and real-time.
Accelerated stability testing studies offer a quick assessment of comparable stability curves for material from before and after a process change — sooner than real-time stability studies. Although working with an especially stable product can be advantageous, it does pose difficulties when trying to degrade that product within a short period. For many biotechnology products, 15- to 30-day accelerated stability studies have been sufficient to evaluate potential degradation pathways and ensure (or discount) material equivalence and compound comparability.
It is often informative to develop an accelerated stability testing protocol that induces degradation pathways similar to those experienced by a given product in real-time storage. Other stress conditions such as vibration, light-exposure, pH changes, and the use of chaotropic agents may also be used to develop an overall stability profile or identify degradation patterns and mechanisms of degradation for a given protein. In most cases, however, stressed temperature conditions provide a well-rounded picture because high temperatures can induce multiple degradation pathways representative of what a product may experience in real time.
One forum attendee questioned whether comparability differences had ever been shown with accelerated stability studies. Several other attendees said that accelerated stability testing had identified differences in their material before and after a manufacturing change. Given that proteins degrade at different rates depending on their structure and product formulation, no set temperature or time conditions are universal.
Long-term or real-time stability conditions are tested under recommended storage conditions of a drug substance and/or product. Such tests ensure that material will meet its end-of-shelf-life specifications and fall within trends for lots manufactured before a change. Long-term stability data are often provided as a postapproval commitment and provided in annual reports.
An important consideration in demonstrating comparability is the distinction between statistical and practical significance. A statistically significant difference can be found in comparing stability curves or a data set, but such differences may be of no practical significance. For example, a change was made in the measurement of cell culture performance parameters that required measurement of total cumulative cell mass to assess specific cell productivity. Analysis of demonstration runs showed a statistically significant difference between three of them. However, when put into the context of cell mass results, no practical significance appeared, and the drug substance resulting from the change was deemed comparable. When developing a prospective comparability plan for assessing the results from a completed comparability study, a manufacturer must carefully consider the potential effects of differences that might be found.
Question 2: Under what conditions is drug-product testing necessary to demonstrate comparability? Forum attendees explored the utility of performing drug-product testing when manufacturing changes are made only upstream from a final drug substance. In some situations, little added value comes from performing extensive drug-product testing in support of a drug-substance manufacturing change, and changes upstream in such a manufacturing process can be fully evaluated by drug-substance comparison testing alone. However, in some cases drug-product testing can help you look for potential effects that would not be apparent with only a drug-substance evaluation. One example is a manufacturing process change that leads to changes primarily in drug-product stability (e.g., by contributing an impurity to the drug substance that does not manifest until subjected to the formulation, conditions, or container–closure system). For instance, metallic activation proteases might have little impact in a drug substance in a frozen solid, crystalline powder, or lyophilized plug state, but they can be quite active in solution.
Evaluation of a drug product assesses its formulation characteristics, quality product attributes, and final product safety — tests that may not be part of a drug-substance comparability assessment. A new impurity identified in drug-product testing might not have been tested for in the drug substance. That was the case with one Novo Nordisk example above, in which an impurity caused by the use of incorrect tubing in drug-substance manufacture was observed only in testing of the final drug product. Such testing also may be necessary for assessment of the “manufacturability” of a postchange drug substance through the drug product process. For situations in which drug-substance manufacturing changes potentially affect a company’s ability to formulate its drug product, laboratory-scale formulation studies have been performed (and included in regulatory submissions) to evaluate the appropriateness of those changes.
Question 3: What changes or circumstances allow for repetitive changes at multiple sites, multiple simultaneous changes, and multitiered changes (e.g., PAS to AR) in reporting through the FDA’s CP mechanism? When plannin
g for a manufacturing process change, companies have the option to submit a comparability protocol (CP) to the FDA describing the planned change and how comparability of the pre- and postchange material will be demonstrated. An approved CP allows a manufacturer to bring postchange product to market faster by eliminating the need for prior FDA-approval following implementation of the manufacturing change. A CP submission describes the change, proposed testing plans, and acceptance criteria that will be used to demonstrate comparability.
The initial submission to gain agreement is a prior approval supplement (PAS), but the follow-up submission may be reduced to a CBE-30/0 or annual report (AR). This allows for more expeditious implementation of a change than from traditional reporting mechanisms, and that can improve resource and material use within a facility. It is of great benefit in coordinating distribution and inventory management for manufacturers with multiple facilities.
Figure 2 shows a timeline for a hypothetical manufacturing change requiring a plant trial and PAS by both the traditional and CP submission mechanisms. When approval is obtained before the plant trial, data are submitted in a follow-up CBE-30 submission, cutting five months from the implementation timeline. CPs have been used for several repetitive changes when a clearly defined method for evaluating the impact of those changes is available.

Repetitive Changes at Multiple Sites: The need to make process and testing improvements is quite pervasive for biopharmaceuticals. The complexity of each molecule and manufacturing process almost guarantees that a wide variety and steady stream of process and analytical testing changes will occur during a product’s life-cycle (16). When a manufacturer has made multiple process changes over a period of years, it may question of what to compare postchange material with. The FDA has recognized that with sequential changes over time, a drift in product characteristics may be observed when comparing A to B, then B to C, and C to D, and so on. Use of retained samples, providing those samples are stable, may be one way to circumvent such drift and evaluate later changes in relation to the original process. Another difficulty with making sequential changes over a lengthy period is that drug substance and product manufactured from different processes and/or different sites may not have been approved in all markets.

Figure 2:
John O’Conner of Genentech noted during his presentation that some manufacturing changes offer no choice but to be implemented at multiple sites. A discontinued filter or packing material may necessitate immediate change at multiple sites. In such a case, qualification of the new supply may be possible at a single site (then simply transferred to the others).
Multiple simultaneous changes and allow comparison back to an original process by implementing all changes at once. Forum attendees agreed that this must be considered in terms of efficiency (How many changes can be covered in a single submission?) weighed against the management of risk for implementing multiple changes. Proposing multiple simultaneous changes is warranted when the outcome is predictable.
It is acknowledged that one change in a process may necessitate a second change to ensure that the end product will be comparable. For example, a site change may also involve column equipment and manufacturing process changes, or a formulation change may necessitate a container–closure change. Assessment of potential product impact from the cumulative affect of such multiple changes should be performed to determine whether a more comprehensive assessment of comparability is warranted.
It is important to consider and compensate for effects that changes to an upstream process steps may have on downstream process steps. One example is the scale-up of cell culture to increase product titer. Modifying cell culture parameters could overload columns in a downstream purification process.
Multitiered Changes: Manufacturing changes that are amenable to a two-level drop in reporting would greatly benefit from using the CP. Potential “double-drop” reporting events would be individual, single-step modifications (e.g., involving a column/buffer or the method for selecting fractions from a column) with clearly defined acceptance criteria. Allison Wolf of Eli Lilly gave an example of using a CP to reduce reporting from a PAS down to an AR for the reprocessing of human growth hormone through the last two steps of a drug substance process. Don’t Be Afraid to Change
An FDA representative polled the audience on how helpful the use of CPs has been. Forum attendees expressed that the reduced reporting mechanism has been very beneficial. One potential shortcoming mentioned suggested that some manufacturers might be discouraged from using a CP from a literal reading of the guidances, which suggests that if comparability data do not meet acceptance criteria, a company must resubmit with a PAS. However, it was the experience of several forum attendees that when acceptance criteria were not exactly met, it did not automatically bump up their follow-up supplements to PAS format. The FDA has been amenable to discussing results that still allowed for reduced reporting in a follow-up supplement. The agency was contacted, and a scientifically justified rationale was presented, so the agency still allowed reduced reporting.
DISCLAIMER
Details contained in this manuscript reflect the discussion that occurred during the January 2005 CMC Forum described above, in addition to the personal experiences of the authors. However, this document does not represent officially sanctioned FDA policy or opinions and should not be used in lieu of published FDA guidance and points-to-consider documents or direct discussions with people at the agency.
ront of this discussion:
aluated in intermediate materials immediately following the process step in which a manufacturing modification was made. A process step further downstream may be selected if a historical database of process-related impurities exists for that step.
g for a manufacturing process change, companies have the option to submit a comparability protocol (CP) to the FDA describing the planned change and how comparability of the pre- and postchange material will be demonstrated. An approved CP allows a manufacturer to bring postchange product to market faster by eliminating the need for prior FDA-approval following implementation of the manufacturing change. A CP submission describes the change, proposed testing plans, and acceptance criteria that will be used to demonstrate comparability.
Repetitive Changes at Multiple Sites: The need to make process and testing improvements is quite pervasive for biopharmaceuticals. The complexity of each molecule and manufacturing process almost guarantees that a wide variety and steady stream of process and analytical testing changes will occur during a product’s life-cycle (16). When a manufacturer has made multiple process changes over a period of years, it may question of what to compare postchange material with. The FDA has recognized that with sequential changes over time, a drift in product characteristics may be observed when comparing A to B, then B to C, and C to D, and so on. Use of retained samples, providing those samples are stable, may be one way to circumvent such drift and evaluate later changes in relation to the original process. Another difficulty with making sequential changes over a lengthy period is that drug substance and product manufactured from different processes and/or different sites may not have been approved in all markets.
John O’Conner of Genentech noted during his presentation that some manufacturing changes offer no choice but to be implemented at multiple sites. A discontinued filter or packing material may necessitate immediate change at multiple sites. In such a case, qualification of the new supply may be possible at a single site (then simply transferred to the others).
Multiple simultaneous changes and allow comparison back to an original process by implementing all changes at once. Forum attendees agreed that this must be considered in terms of efficiency (How many changes can be covered in a single submission?) weighed against the management of risk for implementing multiple changes. Proposing multiple simultaneous changes is warranted when the outcome is predictable.
It is acknowledged that one change in a process may necessitate a second change to ensure that the end product will be comparable. For example, a site change may also involve column equipment and manufacturing process changes, or a formulation change may necessitate a container–closure change. Assessment of potential product impact from the cumulative affect of such multiple changes should be performed to determine whether a more comprehensive assessment of comparability is warranted.
It is important to consider and compensate for effects that changes to an upstream process steps may have on downstream process steps. One example is the scale-up of cell culture to increase product titer. Modifying cell culture parameters could overload columns in a downstream purification process.
Multitiered Changes: Manufacturing changes that are amenable to a two-level drop in reporting would greatly benefit from using the CP. Potential “double-drop” reporting events would be individual, single-step modifications (e.g., involving a column/buffer or the method for selecting fractions from a column) with clearly defined acceptance criteria. Allison Wolf of Eli Lilly gave an example of using a CP to reduce reporting from a PAS down to an AR for the reprocessing of human growth hormone through the last two steps of a drug substance process. Don’t Be Afraid to Change
An FDA representative polled the audience on how helpful the use of CPs has been. Forum attendees expressed that the reduced reporting mechanism has been very beneficial. One potential shortcoming mentioned suggested that some manufacturers might be discouraged from using a CP from a literal reading of the guidances, which suggests that if comparability data do not meet acceptance criteria, a company must resubmit with a PAS. However, it was the experience of several forum attendees that when acceptance criteria were not exactly met, it did not automatically bump up their follow-up supplements to PAS format. The FDA has been amenable to discussing results that still allowed for reduced reporting in a follow-up supplement. The agency was contacted, and a scientifically justified rationale was presented, so the agency still allowed reduced reporting.
DISCLAIMER
Details contained in this manuscript reflect the discussion that occurred during the January 2005 CMC Forum described above, in addition to the personal experiences of the authors. However, this document does not represent officially sanctioned FDA policy or opinions and should not be used in lieu of published FDA guidance and points-to-consider documents or direct discussions with people at the agency.
REFERENCES
1.) CBER/CDER 1996. FDA Guidance Concerning Demonstration of Comparability of Human Biological Products, Including Therapeutic Biotechnology-Derived Products, Food and Drug Administration, Rockville.
2.) EMEA/CHMP/BWP/3207/00/Rev1 2003. EMEA Guideline on Comparability of Medicinal Products Containing Biotechnology-Derived Proteins As Active Substance: Quality Issues. 3.) EMEA/CHMP/3097/02/Final 2003. EMEA Guideline on Comparability of Medicinal Products Containing Biotechnology-Derived Proteins As Active Substance: Non-Clinical and Clinical Issues. 4.) 2003. ICH Q5E: Comparability Biotechnological/Biological Products Subject to Changes in Their Manufacturing Process. Fed. Reg. www.ich.org/LOB/media/MEDIA1196.pdf 70:37861-37862. 5.) 2003. ICH 3QA(R2): Impurities in New Drug Substances. Fed. Reg. www.ich.org/LOB/media/MEDIA422.pdf 68:6924-6925. 6.) 2003. ICH Q3C: Impurities — Guideline for Residual Solvents. Fed. Reg. www.ich.org/LOB/media/MEDIA423.pdf 68:64352-64353. 7.) US FDAGood Manufacturing Practice in Manufacturing, Processing, Packing, or Holding of Drugs: General Code of Fed. Regulations. 8.) US FDACurrent Good Manufacturing Practice for Finished Pharmaceuticals Code of Fed. Regulations. 9.) ICH 2001. Q7A Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients. Fed. Reg. www.ich.org/LOB/media/MEDIA433.pdf 66:49028-49029. 10.) Hermeling, S. 2003. Micelle-Associated Protein in Epoetin Formulations: A Risk Factor for Immunogenicity. Pharmaceut. Res. 20:3. 11.) 2000. ICH Q6A: Specifications — Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products: Chemical Substances. Fed. Reg. www.ich.org/LOB/media/MEDIA430.pdf 65:83041-83063. 12.) 1999. ICH Q6B: Specifications — Test Procedures and Acceptance Criteria for Biotechnological/Biological Products. Fed. Reg. www.ich.org/LOB/media/MEDIA432.pdf 64:44928. 13.) 1966. ICH Q5C: Quality of Biotechnological Products — Stability Testing of Biotechnological/Biological Products. Fed. Reg. www.ich.org/LOB/media/MEDIA432.pdf 61:36466. 14.) EMEA/CVMP/373/04 2005. Guideline on Stability Testing for Applications for Variations to a Marketing Authorisation. 15.) 2004. ICH Q1E: Evaluation of Stability Data. Fed. Reg. www.ich.org/LOB/media/MEDIA415.pdf 69:32010-32011. 16.) Dougherty, J Larger, ES. 2004.Postapproval Changes for Large-Scale Biopharmaceutical Manufacturing: Global Regulatory IssuesAdvances in Large Scale Biopharmaceutical Manufacturing, ASM Press, Washington:555-591.