As regulators become increasingly stringent in demanding a fuller understanding of whole virus preparations, researchers and manufacturers are looking beyond well-known characterization methodologies. Existing technologies for quantifying and characterizing viral preparations such as infectivity assays, quantitative polymerase chain reaction (qPCR), and protein assays provide crucial information but tell only half the story.
We evaluated a unique technology developed by NanoSight Ltd. (www.nanosight.com) for visualizing viruses in liquid suspensions, measuring their approximate concentration, and characterizing their state of aggregation. Information generated by this technology provides complementary data and helps fill in knowledge gaps left after using traditional technologies.
PRODUCT FOCUS: ALL BIOLOGICS
PROCESS FOCUS: DOWNSTREAM PROCESSING AND FORMULATIONS
WHO SHOULD READ: PROCESS DEVELOPMENT, QA/QC, FORMULATIONS, AND ANALYTICAL PERSONNEL
KEYWORDS: VIRAL SAFETY, QPCR, LIGHT-SCATTERING, AGGREGATION, FLUORESCENCE
LEVEL: INTERMEDIATE
Regulatory authorities require some measurements acquired during the development of viral preparations, including the state of aggregation within a viral preparation, the number of viable viruses and total viruses present, ratio of empty to filled capsids, and measurement of residual host-cell proteins. These parameters help define the efficacy of a final product — whether gene delivery vehicle or viral vaccine — as well as its long-term stability, and performance. They also help to predict allergenic reactions in potential patients.

Infectivity Assays
Infectivity assays are the most widely used techniques for estimating live viral titers in a viral preparation. To accurately quantify infectious viral titers, most techniques rely on a single virion infecting a single cell in a culture, with its subsequent replication and infection of surrounding cells causing a plaque to form. The resulting plaques can then be quantified. Although infectivity assays provide crucial information, they do have limitations. So they are generally not used as a stand-alone technique.
Imagine two samples: Sample A has a higher count of PFU (plaque forming units) than Sample B does, but what does this actually mean? The obvious answer is that Sample A has more viable viruses. What is not clear is how many viruses in total are present in each sample. Sample A has a higher PFU count, but what is the ratio of infective to non infective viruses within the sample? It may contain 108 viable particles but may have 1011 total particles with only 1 in 1,000 of those total viruses being viable. Sample B may contain only 107 viable viruses, but if 1 in 10 viruses in the sample are viable, then its total population would be 108 viruses. This is an important distinction to make, especially from a manufacturing and quality point of view. If a company can make a preparation of a specific yield but at higher efficiency, then it can decrease production costs. Moreover, biological responses to nonviable viruses must be understood. If the ratio of infective to non infective viruses can be maximized, then biological performance can be better predicted. For example, analysts may be able to determine the influence noninfective particles have on allergenic responses to a given preparation.
WHAT IS NTA?
Nanoparticle tracking analysis (NTA)is a light-scattering method of nanoparticle analysis. It is increasingly used for determining nanoparticle size through simultaneously tracking and analyzing the trajectories described by individual nanoparticles undergoing Brownian motion in a fluid. The technique centers on a sample analysis module that comprises a small metal housing containing a solid-state, single-mode laser diode (<30 mW, 635 nm). The laser is configured to launch a finely focused beam through a sample of liquid containing a dilute suspension of nanoparticles placed directly above a specially designed optical flat. The sample chamber is ~250 µL in volume and 500 µm deep, and each sample is introduced by syringe through a Luer port. Samples are allowed to thermally equilibrate for 20 seconds before analysis. The beam refracts at the interface between the liquid sample and optical element through which it is passed such that it describes a path nearly parallel to the glass–sample interface (Figure 1).
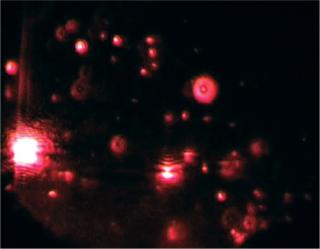
Particles in the beam (which is ~100 µm wide by 25 µm deep) are visualized by a conventional optical microscope aligned normally to the beamax is. It collects light scattered from each and every particle in the field of view. A video of typically 20–60 seconds duration is taken of the moving particles at 30 frames/s (Photo 1). The video is analyzed frame-by-frame with NanoSight’s proprietary analysis program, each particle being automatically identified and located and its movement tracked. Thresholds for particle identification can be user adjusted, as can the camera’s gain and shutter-speed settings, which allows users to optimize the image for a particular sample type. The eight-bit video sequence can be automatically or user-adjusted for images moothing, background subtraction, threshold setting, removal of blurring, and so on to allow particles of interest to be tracked without interference from stray flares or diffraction patterns that occasionally occur with non optimum sample types.
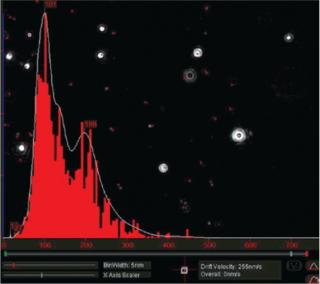
Particles diffusing into the scattering volume are identified and followed for the duration of their presence in the beam — or until they diffuse to within a certain distance of an adjacent particle, at which point tracking ceases, eliminating the possibility of analyzing particle trajectories that cross behind each other (Photo 2). Movements of individual particles are followed through the video sequence, and the mean-squared displacement is determined for each one. From the resulting values, diffusion coefficients and sphere-equivalent hydrodynamic radii can be determined using the Stokes-Einstein equation. Results are shown as a particle size distribution plot (Photo 3).
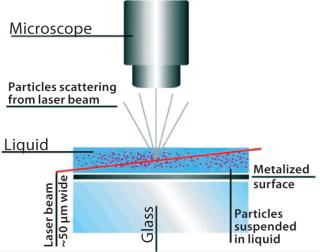
Photo 1:
“font-size:71.429%;”>Photo 1: ()
Photo 2:
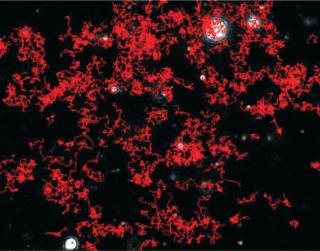
Photo 3:
Another sometimes overlooked reason for disparities in infectious titers comes about with aggregates in a preparation. Sample A may now contain fewer aggregates than Sample B, resulting in a higher PFU count. Both samples could contain the same number of total viruses and the same ratio of infective to non infective viruses, but the degree of aggregation might be different. Infectivity assays are unable to distinguish aggregation because a single virion can infect a single cell and cause a single PFU, whereas an aggregate with possibly more than one viable virus will still infect only a single cell and cause a single PFU. In addition to leading to miscalculation of infectious titers within samples, aggregates within a sample can indicate the long-term stability of a product. So without performing physical characterization on a viral preparation, it is impossible to tell exactly what causes differences in infectious titers:
- Does one sample have a higher titer of viable particles?
- Does one sample have more total particles than another sample?
- What is the ratio of infective to non infective particles?
- Does one sample contain more aggregates than another?
Nanoparticle Tracking Analysis
Nanoparticle tracking analysis (NTA) technology can provide that missing information. As described in the “What Is NTA?” box, the technique simultaneously measures particle-size distribution within a viral preparation and approximate total particle count (infectious and noninfectious virus) of each size. Particle measurements are based on physically counting each virus, which does not rely on biological activity. The technology can visualize light scattered from each individual virus down to a limit of detection (LoD) of ~25 nm in size. With such information, all the above questions can be answered when results are combined with measurements from infectivity assays. In our experience, although NTA provides a good indication of particles per volume, it remains only semi quantitative (especially when dealing with polydisperse samples). Virus Quantification
Quantitative PCR is another commonly used method of virus quantification. The technique works by amplifying DNA (or RNA through a reverse-transcriptase step). The rate at which DNA is amplified depends on the concentration of DNA present. Therefore, analysts can observe the rate of amplification and relate that to the amount of DNA-containing viruses in a preparation. The qPCR technique is advantageous in that it is highly specific and can be used to quantify DNA containing viruses in a harvest material.
Unfortunately, qPCR cannot distinguish the amount of aggregation within a sample preparation. As such, the measurement tells users nothing about the quality of their preparations. In addition, the technique measures only viruses that actually contain DNA or RNA (filled capsids). Many viruses formed in vaccine or gene therapy production contain no RNA or DNA (empty capsids) and thus cannot be measured using qPCR. Also consider infectious and noninfectious particles. It is important to look at the ratio of filled and empty capsids (empty by their nature being noninfectious, although lack of DNA is not the only reason for their loss of infectivity).
Thus, when comparing viral titer provided by qPCR with total viral titers provided by NTA measurement, an analyst can calculate the ratio of filled and empty capsids (despite varying levels of aggregates in a given sample). Dynamic light scattering (DLS) is also commonly used to get size information and some indication of aggregation. The greatest value of NTA in this field is in making it possible to quantify, visualize, and size not only viruses, but also aggregates. Study of Aggregation in Real Time
Our group recently published a critical evaluation of NTA for looking at nanoparticles and protein aggregation (1). One part of our study is included here, in which an IgG formulation is studied at elevated temperature (50 °C for 45 min), and the process of its aggregation is observed. We carried out our work using a NanoSight LM-20system with a temperature control chamber (as shown in the photo on the first page of this article).
At t0, the sample had already been exposed to some heat stress for about 10 min, the time required for the temperature to rise from ambient to 50 °C, and some polydisperse aggregates of about 50–350 nm were detected. At t1, the number of aggregates had increased, with distinct subpopulations becoming apparent ~50 nm, ~100 nm, and ~200 nm. After ~20 min, the number of aggregates rapidly increased. Even though that number had slightly increased at t2, it does not reflect this sudden increase of aggregates because background scattering (visible in the video frame of t2) made particle tracking more difficult for the NTA software. After 35 min of heat stressing, the number of aggregates reached the upper concentration limit of NTA, with especially those ~50 nm in size increasing significantly. After 45 min, very large (>5 µm) aggregates were visible, making accurate NTA size measurements impossible because the light they scattered masked the smaller traceable aggregates (<1 µm). Figure 2 illustrates this study.

Looking to Future Applications
An interesting development in NTA technology involves its ability to visualize fluorescently labeled viruses. In normal operations, images of light scattered from a virus are nonspecific because they don’t depict particles directly but merely the light they scatter. So the technique is best suited for characterizing viruses in purified material. However, labeling viruses with appropriate fluorophores extends the usefulness of this technique into harvest materials, for which it is essential to discriminate viruses from background host-cell debris and proteins. Viruses with appropriately labeled DNA can be discriminated from empty caps
ids and have been detected using NTA. In pure light-scattering mode, both populations would be measured; in fluorescence mode, only the filled capsids would be measured.
Author Details
Corresponding author Vasco Filipe is a PhD student in the Division of Drug Delivery Technology at the Leiden/Amsterdam Center for Drug Research of Leiden University, PO Box 9502, 2300 RA Leiden, The Netherlands; filipev@lacdr.leidenuniv.nl. Wim Jiskoot is a professor of drug delivery in the biologics formulation group, and Andrea Hawe is a postdoctoral researcher, both in the Division of Drug Delivery Technology at the Leiden/Amsterdam Center for Drug Research of Leiden University.
REFERENCES
1.) Filipe, V, A Hawe, and W. Jiskoot. 2010. Critical Evaluation of Nanoparticle Tracking Analysis (NTA) By NanoSight for the Measurement of Nanoparticles and Protein Aggregates. Pharm Res. 27:796-810.