The ultimate goal of recombinant fermentation research is cost-effective production of desired proteins by maximizing volumetric productivity (to obtain the highest amount of protein in a given volume in the least amount of time). Bioprocessing for recombinant proteins using genetically modified organisms requires a stable, high-yielding recombinant culture, a highly productive fermentation process, and cost-effective recovery and purification procedures. Escherichia coli has been a widely used host for expression of recombinant proteins (1). Its advantages lie in the enormous data available on E. coli cell biology and fermentation process development, as well as its ability to produce large quantities of recombinant proteins cost effectively. Early successful production of insulin by Eli Lilly (www.lilly.com) and bovine growth hormone by Monsanto Corporation (www.monsanto.com) validated the versatility and economic potential of E. coli-based therapeutic protein production. Although it cannot be used to produce complex glycoproteins or proteins with multiple disulfide bonds, over the past 20–30 years E. coli has been used to successfully produce numerous products (2,3).
PRODUCT FOCUS: MICROBIAL PRODUCTS EXPRESSED TO INCLUSION BODY FORM
PROCESS FOCUS: RECOVERY
WHO SHOULD READ: PROCESS DEVELOPMENT, MANUFACTURING
KEYWORDS: TFF, INCLUSION BODIES, MICRO- AND ULTRAFILTRATION, CLARIFICATION, DEPTH FILTRATION, PROTEIN REFOLDING, E. COLI
LEVEL: INTERMEDIATE
Recombinant protein expression using E. coli as a host is frequently associated with the formation of intracellular protein aggregates called inclusion bodies (IBs) (4). The volumetric yield is thus a function of both unit-cell concentration and specific cellular protein yield. Optimization of high-cell-density fed-batch fermentation process is a key step in enhancing the volumetric yield of recombinant proteins (5). High-level expression of a protein of interest in IBs facilitates its physical isolation from the cytoplasm at the cost of its native structure. Renaturation to its bioactive form is cumbersome and provides low recovery of the final product. It also accounts for the major cost in overall production of recombinant proteins from microbial systems (6). However, for cases in which a simple, high-yielding protein refolding process is developed for an aggregated recombinant protein, high-level expression of it as IBs provides a straightforward strategy for cost-effective production and purification. Thus, in spite of problems associated with the IBs in E. coli, the system has been extensively used in commercial therapeutic protein production.

MILLIPORE CORPORATION (WWW.MILLIPORE.COM)
Inclusion bodies are dense particles of aggregated protein found in both the cytoplasmic and periplasmic space when E. coli expresses foreign proteins at high levels. IB size varies 0.5–1.3 µM, and IB protein aggregation may be either amorphous or paracrystalline depending on localization (7). In many cases, IBs constitute 20–50% of the total cellular protein in a bacterium.
In addition to heterologous proteins, IBs contain very small amounts of host protein, ribosomal components, and DNA/RNA fragments. It has also been reported that the presence of contaminants in isolated IBs is mainly due to incomplete purification of them following cell lysis (8), but it could result from accidental trapping during the aggregation of polypeptide into IB form. IBs often contain the overexpressed protein almost exclusively, and such aggregation has been reported to be reversible (8).
Formation of inclusion bodies thus facilitates the easy isolation and recovery of expressed proteins in a denatured form. Because IBs have high density (∼1.3 mg/mL), they are easily separated by high-speed centrifugation after cell disruption. The most efficient process for complete cell lysis is high-pressure disruption. Lysozyme is used rarely in large-scale processes because of its cost and regulatory concerns about animal-sourced material. Homogenization is more often performed using mild detergents, although lysozyme is still used in some processes developed some time ago.
Further purification can be achieved by washing with detergents, a low concentration of salt, and/or urea. With proper isolation and washing, IBs of >95% purity can be prepared from E. coli. Some studies indicate that sucrose-gradient centrifugation can provide very pure IB preparations; however, preparative gradients are not scalable or cost effective for production. Ultrafiltration with membranes of different pore size has also been used for IB isolation from E. coli cells (9).
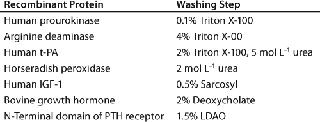
Purification Using Membrane TechnologyIB isolates collected after maximum cell disruption are usually quite homogeneous, and proteins can be renatured directly after solubilization without further purification. However, both proteinaceous and nonproteinaceous IB contaminations may interfere with subsequent renaturation of a recombinant protein. In such cases, the IBs can be further purified by additional washing steps using EDTA, low-concentration denaturants such as urea or guanidium hydrochloride, and detergents such as Triton X-100, deoxycholate, octylglycoside, and sarcosyl (Table 1). In addition, IBs can be purified by gel-filtration chromatography. But it is limited as a method for IB purification because of issues with scalability, extended operation times, limitations of product loading, and operation costs. So the most common methods adopted for industrial IB purification are centrifugation and membrane technology (9).
Table 1: Buffer additives for washing of inclusion bodies (11)
After being suspended in washing buffer to remove contaminants, IBs can be centrifuged to form pellets, which are recovered for further processing. IB purification often requires several washing cycles, however, with recovered pellets resuspended in washing buffers. Continuous pellet recovery from a centrifuge and resuspension in wash buffer can lead to product loss. Nevertheless, high-volume centrifugation may be limited by the achievable rotation speed and bowl capacity.
Membrane-based technology for IB washing and subsequent purification avoids such limitations and offers scalability (10). After resuspension in wash buffers, IBs can be continuously washed in diafiltration mode using tangential-flow filtration (TFF), then concentrated to a desired volume after washing. Because most IBs are dense, retractile materials insensitive to shear, they can be processed easily using membrane-based TFF (Figure 1).
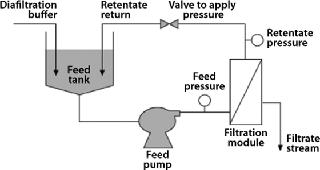
Common formats used for TFF include flat-plate thin-channel cassettes, and spiral-wound and hollow-fiber filters. Figure 2 shows the basic flow path of each. Polyvinylidene fluoride (PVDF) and polyethersulfone (PES) membranes are well suited for IB washing and purification applications because of their chemical resistance and robustness. TFF devices may also use different screens (Figure 3). An open-channel type TFF device is well suited for this application.
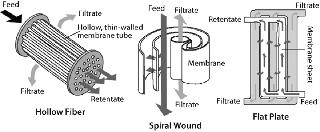

Most TFF devices are limited in their capacity to handle suspended solids in processing solutions. It may be difficult to handle >25% packed suspended solid in any given solution using TFF. Nevertheless, a well optimized and controlled microfiltration operation with permeate control (HR-TFF) can handle 20–25% suspended solids. Depending on the size and tightness of IBs, 0.1-µm microfiltration or 1,000-kDa (V-screen) ultrafiltration could be ideal for processing them. High cross flow but low transmembrane pressure (TMP) could be beneficial to the effectiveness of IB washing because of high recirculation.
Case Study 1We performed a comparative analysis of two different washing methods: IB washing by centrifugation and IB washing by TFF. We considered >90% purity of IB (as measured by densitometric analysis of SDS-PAGE) as our benchmark and used buffer containing 1-M urea for IB washing. Inclusion bodies of chimeric dengue multiepitope protein (∼45 kDa) expressed in E. coli were isolated by cell lysis. In one study, IBs were pelleted by centrifugation, followed by two centrifugal washings at 13,000g. In the other study, cells were lysed in buffer containing 1-M urea, and the suspended solids were adjusted to 4% in wash buffer. IB washing was performed in this case using a Millipore Pellicon 2, Biomax, 1,000-kDa, V-screen, 50-cm2 TFF device operating in constant-volume diafiltration (2-DF volume) mode.
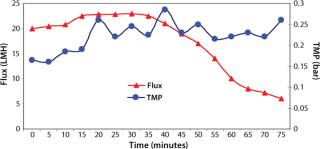
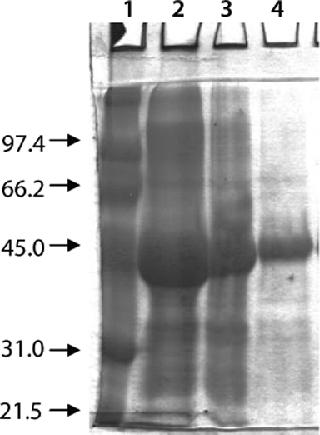
Washing by centrifugation resulted in 18% loss of protein, mostly due to loss of IBs during wash cycles. Some nonpelleted IBs were properly lost in centrate. Resuspension in wash buffer was cumbersome, and washing was not effective enough to provide the desired purity of IB. Alternatively, cell lysis in buffer containing 1-M urea followed by washing using TFF gave >95% recovery of protein by two diafiltrations. An average 0.2-bar TMP was maintained throughout the process, and postwashing IBs were concentrated 2× before solubilization (Figure 4). SDS-PAGE analysis reveled higher purity of the postwash IB (after diafiltration) compared with prewash IB (Figure 5). There was no IB passage in TFF, but we found some protein loss of protein (Figure 5) due to 1-M urea exposure over time. Our later experiments confirmed the same, and later we achieved similar IB purity without any significant loss of protein by washing with buffer containing 700-mM urea instead. Considering the processing time, buffer use, protein recovery, and purity, we found that method to be economical (unpublished data).
Protein Refolding: Our strategy to recover bioactive protein from inclusion bodies involved four steps: isolation and purification of inclusion bodies from E. coli cells, solubilization of protein aggregates, refolding of solubilized protein, and purification. Refolding is the most crucial step and needs careful attention for high protein recovery. Most aggregation that leads to low protein recovery is attributable to suboptimal refolding procedures. Because protein aggregation leading to IB formation is highly specific, >90% IB purity can be achieved using optimal washing procedures, which helps reduce the number of steps in further downstream purification.
In general, soluble proteins are refolded into their native state after removing chaotropic agents or other salts by dialyzing the proteins in buffers containing reducing and oxidizing agents. Purification is carried out either before renaturation (at denaturing conditions) or after refolding of a solubilized protein. Refolding followed by purification is generally preferable because some high-molecular aggregates can be copurified along with the contaminants in a single step.
Despite several protocols available for IB protein solubilization and refolding, the overall recovery of bioactive proteins is often very low (12,13). Most protein molecules that fail to achieve a thermodynamically stable state will either aggregate or precipitate during refolding. Efficiency of protein refolding and product recovery (∼15–25%) is lower than for other unit applications (14). But significant process improvements have been made over the past few years, resulting in 60-70% recovery from this unit operation (15,16,17). Multimerization of a protein during refolding causes haziness/cloudiness leading to product precipitation. It is essential to clarify a refolded protein solution to remove aggregates that can interfere with or affect the performance of subsequent downstream unit applications such as chromatography or ultrafiltration (10). The level of suspended matter in all cases (which varies by method) will cause rapid downstream fouling with a significant impact on cost.
Depth Filtration for Clarification of Refolded Protein: Normal-flow filtration and/or dead-end filtration (clarification by depth filtration) can remove protein aggregates. The challenge confronting process engineers here is to arrive at an integrated clarification and sterile filtration train that minimizes overall production costs. In many cases, clarification of a protein refolding solution involves several serial filtration steps. A need for multiple separations arises because of the broad range of particle sizes and types present in process streams. Use of a multigrade depth filter combined with a membrane filter consolidating these multiple serial filtration steps into a single operation is now common practice for removal of protein aggregates. Although a few manufacturers are still using centrifugation, the practice has rpm limitations, requiring subsequent membrane filtration for complete removal of protein aggregates before a chromatography operation (18).
Protein precipitates/solids exhibit poor filter-cake properties; very thin deposits exhibit high flow resistance. The capacity of a clarifying depth filter therefore depends highly on the accessible internal surface area available for particle deposition (19). For a given media type and grade, capacity increases linearly with thickness or bed depth. Moreover, often in biological suspensions the particles span a broad range of sizes. So to maximize capacity filter media must be structured to accommodate them in proportion to their number. So a gradient pore structure or media density is optimal. Coupling two cellulosic pads within a multilayer composite creates a graded pore structure from front to back, which helps expand the effective pore size range by as much as several orders of magnitude (20).
Placing microporous membrane layers downstream of the cellulosic pads in the design of stacked-disk prefilters delivers two important advantages: It serves as a final barrier to trap small particles and colloids that would otherwise foul a trailing sterile filter, and the membrane contributes a modest hydraulic resistance to the filter (significantly improving feed-flow distribution and, consequently, media use). All vertical stacked-disk designs inherently force a greater percentage of feed flow to the lower sections of a filter column, which are under higher hydrostatic pressure. Addition of a microporous membrane to a depth filter helps minimize that vertical pressure gradient to make more even and effective use of all available filter area (21).
Our internal study results suggest that when applied to clarification of refolded protein solutions with product aggregates or precipitates, graded-density depth filters using an additional microporous membrane depth will ordinarily develop little pressure buildup but remarkably improve downstream sterile filter throughput. Such depth filters could reach a process capacity of >500 L/m2 while never exceeding a pressure differential of 5 psid. If the resulting filtrate require sterile filtration, then the throughput on the trailing sterile filter could be >3,000 L/m2.
The aggregate or precipitate removal achieved by graded density depth filters is the consequence of two distinct separation mechanisms. The first is mechanical sieving, in which particles are captured within the prefilter because of their physical size and/or orientation. In a depth filter, the path through the media matrix is tortuous and varies greatly in its effective pore dimensions, which provides ample opportunity for particle retention by entrapment. Typical of most filters, the outward effect is a build-up in pressure differential along the direction of flow. The second particle-capture mechanism for such filters is adsorption: the physical–chemical binding of particles to the filter medium. This process is aided by a net positive surface charge on depth filter media. Colloidal particles in a biological suspension (e.g., cell wall fragments, agglomerated proteins, and polynucleic acids) exhibit a net negative electrostatic charge on their surfaces at typical nearneutral pH conditions. A cationic charge on depth filter media allows such particles to be captured when otherwise they might be small enough to escape retention. This electrostatic retention mechanism allows a charged depth filter to be effective in removing particles well below its smallest pore size and thereby provides additional protection to a sterile filter (22,23).
However, that capture mechanism does not reveal itself as a pressure buildup on a prefilter. Instead, users must carefully monitor filtrate quality to detect the point at which all available adsorption sites within a filter have been used. This can be done either by monitoring filtrate turbidity in relation to process throughput or measuring the capacity of a downstream sterile filter in situ or offline. Depending on the properties of the feedstream (and the consequent relative contribution of the two separation mechanisms described above), two distinct types of behavior may be observed for graded-density charged depth filters (Figure 6). When the dominant capture mechanism is adsorption, filtrate turbidity will be seen to rise markedly as the prefilter’s capacity is reached while the pressure differential across it rises more slowly (a “breakthrough-limited” profile). When the dominant capture mechanism is mechanical sieving, the pressure differential will be seen to rise markedly across the prefilter as its capacity is reached, and filtrate turbidity rises more slowly (a “pressure-limited” profile). Testing will determine which applies — and therefore whether a prefilter pressure limit or breakthrough limit should be applied in overall filter train sizing.
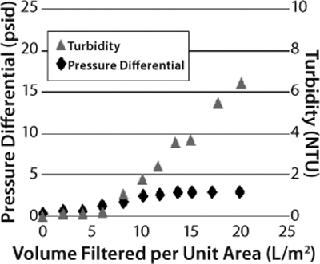
A well-optimized in-vitro protein refolding step may show 5–50 NTU turbidity. Based on in-house standards, the quality of filtrate from a depth filter might be 10% of the feed turbidity. It is essential to monitor filtrate quality, not only for filtrate clarity, but also product recovery. If the target protein in refolding buffer has a negative net charge, then it could bind to the depth filtration matrix. Such situations could be controlled by using high-salt buffers.
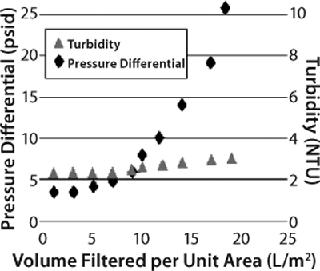
Case Study 2We evaluated three different Millistak+HC graded-density charged depth filters for clarification of recombinant granulocyte colony stimulating factor (G-CSF) expressed in E. coli. The G-CSF was purified from IB form and refolded by an oxidative method to produce a mixture of soluble and aggregated protein. The refolded solution appeared cloudy, having a turbidity of 46 NTU. We used miniature capsules (23 cm2) containing two layers of depth filtration media — namely Millistak+A1HC (60DE 0.1-0.4 µm, 75DE <0.1 µm), RW01 0.1 µm), Millistak+B1HC (50DE 0.2–0.7 µm), 75DE <0.1 µm, RW01 0.1 µm), and Millistak+C0HC (30DE 1.0–3.0 µm), 60DE 0.2–0.4 µm) — at a constant flow rate of 6.4 mL/min (165 LMH). And we considered a pressure differential of 20 psi or a 10% NTU breakthrough to be the operation limit.
The Millistak+A1HC filter showed faster pressure breakthrough, and the Millistak+C0HC filter showed faster turbidity breakthrough, requiring higher filtration area to achieve desired throughput and filtrate quality. The Millistak+B1HC filter exhibited high capacity (150 L/m2), with filtrate having a pooled NTU of <5.0 (Figure 7). The A1HC filter plugged faster because of its tighter pore structure building resistance, whereas the C0HC filter could not retain the fine particulates/precipitates (colloidal material) because of its more open pore structure and lack of 0.1-µm filter after the depth filter layers.
E. coli continues to be a useful production host for large-scale expression and purification of nonglycosylated recombinant proteins. Successful production of heterologous bioactive protein from this bacterial cell line requires a multidisciplinary approach that considers the effects of cell and molecular biology, fermentation process development, high-throughput purification, and stable formulation. However, commercialization success mostly depends on volumetric productivity of a purified protein. In this context, highly productive fermentation processes and high-throughput purification become the most important bioprocess engineering parameters for successful production of therapeutic proteins from E. coli.

With the cell biology information available and developments of high-density fed-batch culture, it has been possible to achieve very high cell densities with E. coli. This has in turn helped reduce the size of production reactors, which has improved the overall costs of production. Cell densities up to 200 g/L have been achieved. With protein expression at such high cell densities, volumetric yield will be very high. Coupled with good understanding of a protein’s structure, aggregation behavior, and its nature in IB form, this has helped improve volumetric yields.
Developments of new and novel protein refolding procedures have not only improved the recovery of bioactive proteins from E. coli, but also provided new information about their behavior at high concentration. Using these novel solubilization and refolding procedures, recovery of bioactive protein could be improved considerably. With interactive analysis of the effects of high-cell-density fermentation and IB solubilization, the overall yield can be improved, and such methodologies can be applied to bioprocess development.