Due to the molecular complexity of monoclonal antibodies (MAbs) and potential impurities in cell culture media before purification (host-cell proteins, DNA, media components) (1), subsequent downstream operations must consistently and reproducibly purify products to ensure safety and efficacy.
The latest member of GE Healthcare’s MabSelect family is called MabSelect SuRe LX (2). As Table 1 shows, it has been developed using the same highly cross-linked agarose base matrix and protein A ligand as for other MAb affinity resins (Table 1). Here, we provide an overview of a novel system for MAb capture from a user’s perspective.
Table 1: Comparing the specifications for MabSelect SuRe and MabSelect SuRe LX protein A adsorbents; a higher dynamic binding capacity is observed (using a longer residence time) with MabSelect SuRe LX (MabSelect media, technical information, GE Healthcare).

Interaction involves fragment B of protein A, which establishes a highly selective interaction with IgG and is composed of three alpha helices. As Figure 1 shows, side-chain residues on the binding surfaces of two helices contribute to most of the interactions with IgG (3). Using the same product-contact materials as previous MabSelect resins provides some familiarity to quality control laboratories that have developed, qualified, and validated protein A assays used to evaluate ligand leakage. Extensive analytical validation is unlikely to be required if the new resin is considered as an alternative for commercial production (4).
PRODUCT FOCUS: ANTIBODIES
PROCESS FOCUS: DOWNSTREAM PROCESSING
WHO SHOULD READ: PROCESS DEVELOPMENT AND MANUFACTURING
KEYWORDS: PURIFICATION, PROTEIN A, RESIN, HCIC, PROTEIN A MIMETICS, ALTERNATIVES
LEVEL: INTERMEDIATE
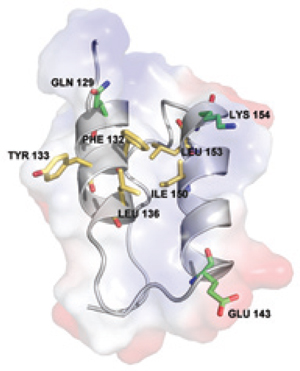
Figure 1:
The ligand density of this new adsorbent has been increased and optimized, with reported dynamic binding capacities (with polyclonal and monoclonal human IgGs) of ≥60 g/L (5). One potential limitationis that longer residence times during column loading (>6 minutes) are required. This time is to permit interaction with protein A coupled to the inner surfaces of the beads. Linear flow rates could be increased for other steps of the chromatographic process (such as equilibration, washing, regeneration, etc). Therefore the slower flow rates during product loading are unlikely to have a significant impact on process capability at production scale.
Reported data indicate that the adsorbent’s binding capacity is independent of MAb feed concentration, with comparable resin binding capacity at feed concentration of 1–10 g/L IgG expressed in a CHO cell supernatant. The concentration independent performance is of interest, particularly if higher-titer MAbs are expressed for commercial processes and smaller-diameter columns are used in downstream processing steps. Pressure flow characteristics and alkaline stability of the protein A is also reportedly maintained, with an extended lifetime for MabSelect SuRe LX media (80% of the dynamic binding capacity reported after 150 cycles with cleaning buffer consisting of 0.5M NaOH) (6). With an increased density of protein A ligand, analysts might anticipate an increase in leached ligand, particularly during the first few affinity cycles. Low levels of leached protein A (10–15 ppm) have been reported. These levels are higher than those typically observed for other affinity adsorbents in the MabSelect range (5–10 ppm). Nonetheless, any minor potential increase of leached ligand is likely to be removed during subsequent ion-exchange steps typically included in a platform purification process.
Host-cell protein (HCP) levels in the eluate fraction are reported to be slightly higher compared with other MabSelect adsorbents. Such levels are potentially a result of the higher loading of antibody and the subsequent higher concentrations of MAb in the eluate. The increased residence time during column loading may also provide additional time for HCPs to diffuse into the pores of the beads. Furthermore, HCP levels in cell culture harvest may vary depending on the variable levels of residual proteins in a bioreactor, so the HCP levels in the MabSelect pool may vary considerably. Therefore, HCP clearance should be evaluated case by case.
Modifications within the operating ranges of buffer (e.g., pH and conductivity) during the primary capture step (e.g., column washing and elution buffers) may vary impurity levels in the product pool. HCP levels should be assessed during process development and/or characterization. Platform approaches for MAb purification have shown that impurity levels may be reduced to appropriate values (low ng/mg) following subsequent downstream steps (7).
Interactions: Increasing protein A ligand density on chromatography beads may appear to be an obvious modification to improve adsorbent capacity and performance, but this approach may not be altogether straightforward. Interactions between MAb and protein A domains are determined by taking into account not only the properties of the ligand, but also the chemical nature of the solid support used (8) and molecular spacers that connect the ligand to a bead.
Interactions of protein A with IgG are thought to be stabilized predominantly through hydrophobic interactions with aromatic protein A residues Phe 132 and Tyr 133 (8). Such reports agree with the structure of the region between the CH2 and CH3 domains of the Fc fragment, which forms a hydrophobic pocket on the surface of the protein (Figure 2). A number of relatively simple synthetic protein A ligands have been developed using a triazine scaffold and two functional groups that mimic the structure and orientation of the Tyr and Phe amino acids (9) and bind to IgG when coupled with solid supports. Other data suggest, however, that the role of those hydrophobic residues may have been over-emphasized and that other more subtle electrostatic interactions contribute to the binding surface when the proteins interact (8).
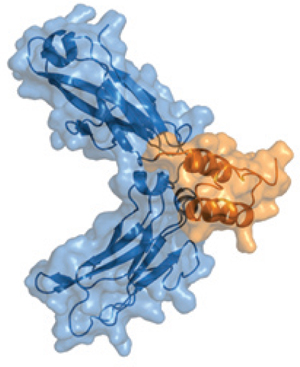
Figure 2:
A MAb purification process consisting of an affinity capture step (e.g., protein A) is often considered the most costly step in a platform process because of the capital investment needed to buy chromatography media (10). As a result, several alternative downstream technologies have been evaluated for the commercial-scale purification of antibodies.
Companies also have developed and evaluated methods for antibody purification without the need for a protein A capture step. Although a detailed discussion is beyond the scope of this article, some frequently reported primary capture steps include the use of mixed-mode chromatography resins (hydrophobic charge induction chromatography, HCIC) and synthetic protein A ‘mimetic’ resins. Such resins typically consist of mixed-mode heterocyclic ligands.
Adsorption of antibodies to HCIC resins can occur through a combination of mixed interactions (hydrophobic and ionic) — often without high salt concentrations and product eluted from the adsorbent using low pH buffer (11). Although this technology has been effective, mixed-mode chromatography generally tends to have lower process yields and less desirable impurity profiles due to increased nonspecific binding (12). Furthermore, overall quality profiles of an eluted product can vary depending on impurity levels in a cell culture harvest material. So HCIC adsorbents may be more difficult to apply as a MAb platform process.
Researchers have developed synthetic protein A ligands based on the hydrophobic IgG binding domain of protein A using techniques such as protein engineering, phage display, and synthetic chemistry (11). Initial studies showed that capture of antibodies using a protein A synthetic resins could yield comparable product quality and throughput (13). More comprehensive studies, however, showed that synthetic ligands generally reduce selectivity, affinity, and a degree of nonspecific adsorption compared with biological ligands such as protein A (11,14,15). Some studies demonstrated differences in selectivity between two antibodies (15), resulting in the need for operational and/or process modifications for similar MAb products.
Synthetic ligand adsorbents are potential alternatives to protein A, but they may be challenging to implement as part of a platform process for commercial antibody purification. Although the capital expenditure required for MabSelect resin may not be insignificant and primary capture may require the highest level of capital investment for a typical Mab platform process, it may represent only ∼3% of total manufacturing costs (10).
Furthermore, if the new generation of protein A resins are considered (e.g., MabSelect SuRe and MabSelect LX brands) with greater ligand density and stability permitting ≥150 reuse cycles (6), then the associated contribution of the resin to the cost of goods per cost per batch is reduced. Such cost then may be comparable with those of single-use virus removal filters (depending on column/batch size) often used in MAb manufacturing. Replacing protein A with a less expensive ‘alternative’ could reduce consumable costs, but it may not have a significant impact on cost of goods because of resulting productivity losses (10).
Options for the Next GenerationThe potential decline of the dependence on affinity chromatography for the primary capture of MAbs may have been quelled — for now. But synthetic-ligand, mixed-mode, or protein A affinity adsorbents may need to evolve further still to meet ever increasing demands. Some options for a next-generation adsorbent development could include higher ligand density, alternative ligand orientation and accessibility, changes in bead and pore size, or modified support matrices that demonstrate increased size and potential mass transfer (15). Although protein A affinity adsorbents may be here to stay, investment in subsequent adsorbent evolution for the primary capture of antibodies may be driven by throughput requirements, process economics, and the number of monoclonals progressing to commercial manufacture.
Author Details
At the time of article preparation, author affiliations were as follows: Piranavan Thillaivinayagalingam as senior scientist, Keara Reidy as QC bioanalyst, Anna Lindeberg as technical services lead, and corresponding author Anthony R. Newcombe as purification lead, all at Pfizer Biotechnology Ireland;
REFERENCES
ol. Biomed. Life Sci. 848:48-63.