Endoproteases specific for cleavage of peptidyl bonds on the C-terminal side of lysine residues (e.g., Lys-C) are produced from a number of bacterial species, including Achromobacter lyticus (1), Pseudomonas aeruginosa (2), and Lysobacter enzymogenes (3). The Achromobacter protease 1 (API) protein has been substantially characterized (4,5,6) and shown to be a resilient enzyme that can specifically cleave after lysine residues under a wide range of buffer conditions, including high concentrations of denaturing agents such as urea and sodium dodecyl sulphate (SDS). Those properties make this protease particularly useful for peptide mapping (3) and in-gel digestion (7) applications.
PRODUCT FOCUS: PROTEINS
PROCESS FOCUS: DOWNSTREAM PROCESSING
WHO SHOULD READ: R&D, PROCESS DEVELOPMENT, ANALYTICAL, AND MANUFACTURING PERSONNEL
KEYWORDS: LYSOBACTER ENZYMOGENES, LYS-C, ANIMAL-FREE MEDIA, PLANT-BASED EXTRACTS, DISPOSABLE BAG FERMENTORS, PROTEASE PURIFICATION, SEQUENTIAL CHROMATOGRAPHY, ENZYME CHARACTERIZATION
LEVEL: ADVANCED
Starting with the Lysobacter enzymogenes American Type Culture Collection (ATCC) strain 27796, we developed an animal-free media composition by optimizing both the plant-based protein extract source and sugar type. Then we scaled up the Lysobacter culture to 40 L using a Cellexus fermentor with disposable bag technology. Various purification schemes for Lys-C have been published involving several chromatography steps and various precipitation techniques (4, 8, 9). All those purification schemes take multiple days and involve numerous concentration/dialysis steps as well as cold temperatures to give good protease recovery with little loss due to self digestion. Here, we combine the different attributes of four chromatography media to elicit rapid purification of the endoprotease Lys-C from culture supernatant. By sequentially loading the elution fractions from one column directly (or with minimal sample handling) onto the next column in the series, we reduced the purification time and therefore self digestion of the protease compared with other methods. Our process yielded pure Lys-C protease with specific activity towards lysine residues, a low endotoxin level, stable storage characteristics, and good tolerance to freeze–thaw cycles. We produced a final Lys-C preparation without any contact with animal products, and the process can be scaled to produce gram quantities of the enzyme.

ALEXEY STIOP WWW.ISTOCKPHOTO.COM
A complete amino acid sequence of the isolated Lys-C protein was determined by cloning and sequencing the gene from Lysobacter strain ATCC 27796. We identified a new isoform of Lys-C that shares mixed identity with both the Lep A and Lep B genes already published for Lys-C proteases derived from Lysobacter (9, 11).
Materials and MethodsCulture Conditions: We grew Lysobacter enzymogenes subspecies enzymogenes (ATCC 27796) cultures in an animal-free medium containing 1% HiVeg plant extract from HiMedia Laboratories (www.himedialabs.com), 1% glucose, 0.01% of each mono- and dibasic potassium phosphate, and 0.02% magnesium sulphate. All other tested media substitutions are indicated in the results section, below. We used shake-flask cultures at 50-mL and 1-L volumes to generate a 1-L fermentor inoculum. The initial OD600 measurement for each shake flask culture was 0.05 ± 0.01. Cultures grew at 30 °C with 225 rpm shaking, and final OD600 measurements of 3.5–4.5 and 2.5–3.0 were achieved after ∼24 hours for the 50-mL and 1-L cultures respectively.
The fermentor medium’s carbon source was supplemented to a level of 2% (w/v) glucose. Fermentation cultures were initiated at a starting OD600 of 0.06–0.07 and reached a final OD600 of 4.5–5.5 after ∼72 hours at 30 °C with 8-L/min aeration. Fermentation was carried out in a Cellmaker Lite 2 from Cellexus Inc. (www.cellexusbiosystems.com), using disposable CellexusBag technology for aeration and mixing. Our assembly followed the manufacturer’s recommendation using filters for sterile gas exchange and media loading as well as a sterile sample/injection port assembly. All sterile connections to the fermentor bag were made in a laminar flow hood.
Enzyme Purification: From the 40-L fermentation culture, we clarified 36 L using batch centrifugation in a Beckman/Coulter Avanti J-20 XP centrifuge (www.beckman.com) with a JLA 8.1000 rotor at 15,900g for 15 minutes. Then we collected the culture media and passed it in 11-L batches through a 0.45-µm filter from Pall Corporation (www.pall.com) onto a BPG-200 column packed with 2 L of DEAE-Sepharose media from GE Healthcare Bio-Sciences AB (www.gehealthcare.com) using a 620S peristaltic pump from Watson-Marlow Bredel (www.watson-marlow.com). The flow-through volume was collected and the active protease precipitated with 70% (w/v) ammonium sulphate for an hour at 4 °C with stirring.
Following centrifugation of each batch using the same centrifuge and rotor at 15,900g for 30 minutes, the protein pellet was solubilized in 1 L of 1-M NaCl and 20-mM Tris at pH 8.0, then loaded onto an XK 50 column from GE Healthcare packed with 100 mL of PPA Hypercel resin from Pall equilibrated in the same buffer. We allowed a residence time of 10 minutes (with a 10-mL/min flow rate) to ensure complete binding before washing with three column volumes of buffer (1-M NaCl and 20-mM Tris at pH 8.0).
Lys-C was batch eluted with 8-M urea and 20-mM Tris at pH 8.0. A single peak containing protease activity (as determined by colorimetric assay) was pooled and loaded at 5 mL/min directly onto a reverse-phase Tricorn column from GE Healthcare packed with 9 mL of Resource 15 reverse-phase media, also from GE Healthcare. After the column was washed with two column volumes of 20-mM Tris at pH 8.0 and 10% acetonitrile, a 10–25% linear gradient of acetonitrile was initiated. Fractions containing protease activity were pooled, diluted twofold with 20-mM Tris at pH 8.0, and loaded directly onto a ϖ-AminoHexyl Sepharose column at 2 mL/min (an XK 16 column from GE Healthcare Bio-Sciences AB) packed with 8 mL of ϖ-AminoHexyl-sepharose media from Sigma-Aldrich Canada Ltd. (www.sigmaaldrich.com). The column was washed with 20-mM Tris at pH 8.0 and then eluted with a 0–700 mM linear gradient of NaCl. We observed a single peak that contained Lys-C protease activity and pooled the fractions containing the highest protease activity, then quantitated that activity by colorimetric assay. Samples were then aliquotted for storage at −20 °C.
Colorimetric Protease Assay: We monitored the Lys-C enzyme reaction rate against the substrate Ac-Lys-pNA (Bachem Catalog #L-1045, www.bachem.com) at 405 nm over a period of 30 minutes using a SpectraMax Plus 384 microplate reader from Molecular Devices (www.moleculardevices.com). Each 200-µL Lys-C reaction included 185 µL of 180-mM Tris-HCl at pH 9.0, 250-µM Ac-Lys-pNA, and 15 µL of test sample. We measured the reaction rate as a change in 405-nm absorbance over time (mOD/min) and extracted the maximum slope from the resulting curve.
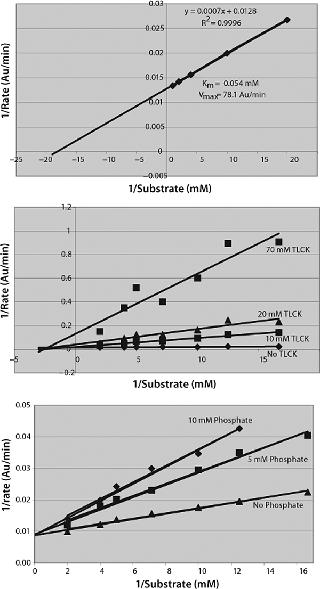
Lys-C Characterization Assays: We determined Lys-C proteolytic activity at different pH values (4,5,6,7,8,9,10,11,12,13) using the colorimetric assay as above. The substrate at 250 µM in 200-mM Tris was mixed with 5 µL of Lys-C. (Various pH values were generated by mixing 200-mM Tris-HCl with 200-mM Tris-Base, and for the very high pH values 10-M NaOH was added.) We assessed protease activity at each pH value by noting the initial rate of the proteolytic reaction. We similarly determined the effects of other additives (urea, guanidine hydrochloride, NaCl, acetonitrile, and SDS) by preparing a series of buffers containing those additives and then adding a stock solution of substrate, then initiating a reaction by addition of the Lys-C. To determine the enzyme kinetics, we prepared various buffers containing different amounts of the colorimetric substrate. We estimated the Michealis constant (Km) and Vmax values from a Lineweaver-Burke plot. Repeating this analysis with increasing concentrations of different inhibitors helped us determine the inhibition constant (Ki) for competitive inhibitors.
Cloning and Gene Sequencing: We assembled the gene sequence encoding Lys-C from L. enzymogenes (ATCC 27796) from three independent but overlapping fragments amplified by polymerase chain reaction (PCR). For the first fragment, a forward primer (5′-GGCCAGTGCAACGTCGAC GTGGTCTGCC-3′) was designed based on an N-terminal amino-acid sequence (GVSGQCNVDVVCP) recovered from the purified protein. This was combined with a reverse primer (5′-GTGCGGTTGTGCAG GTTGTAGAC-3′) designed to a region conserved within the C-extension domain in both lepA (Genbank Accession AB045676) and lepB (Genbank Accession AB094439) genes from Lysobacter species IB-9374. That fragment provided the DNA sequence of the mature protease coding sequence and a portion of the C extension.
To isolate the N-terminal prepro sequence, we designed a reverse primer within the recovered sequence of the first fragment (5′-GATAGTTCCAATACACCACGATG-3′) and paired it with a forward primer (5′-CTTCAAACGCACGCTGTAGGGGAAG-3′) designed from the published lepA promoter region that spans the 35–10 region. The third C-terminal fragment was isolated by chromosome walking using a single specific primer to the C-extension region (5′-CGATCTACCACACCTACAAGAG-3′).
Each PCR used 50 ng of sonicated Lysobacter genomic DNA as a template and included 1× Q-solution from Qiagen (www.qiagen.com) in the reaction to facilitate amplification from the high-percentage guanine–cytosine DNA of Lysobacter. We cloned the PCR products into a pGEM-T vector for sequencing. DNA sequencing of the lys-c gene on both strands using the dideoxy chain termination method (12) performed at the University of Calgary Core DNA Sequencing Laboratory (www.ucalgary.ca/dnalab). The resulting DNA sequence and its deduced amino-acid sequence were analyzed using Vector NTI software (www.invitrogen.com).
ResultsStarting with Medium A, an animal-based composition for Lysobacter published by Chohnan et al. (9), we tested plant-derived protein extract sources to find a suitable replacement for milk casein and beef extract components. The nitrogen component was found to greatly influence cell densities achieved with another Lysobacter species, lactamgenus (13). To optimize the protein extract for Lysobacter enzymogenes growth and Lys-C enzyme production, we tested several different sources of plant-derived protein extracts at a level of 1%; including HiVeg extract from HiMedia Laboratories and the LucraTone series of hydrolysates (e.g., broadbean, pea, soy F, soy P, wheat, lupin) from Millipore Corporation (www.millipore.com/processdv/pd3/lucratone).
We included yeast extract in our study as a non-plant comparison. Test cultures for each extract were run in parallel, and Lys-C activity was monitored over several days of growth (Figure 1-LEFT). Although all extracts supported similar cell growth, Lys-C activity levels in culture supernatant were ∼2.0–2.5× higher with the yeast and the lupin plant extracts. Although the HiVeg extract with 1% sucrose gave one of the lowest levels of Lys-C activity <0.5 mOD/min), but when it was combined with sugar optimization (described below), that activity was raised to >3.0-3.5 mOD/min (Figure 1-RIGHT).

We optimized the carbon source for our medium from the starting condition of 1% sucrose (9). Based on published findings for Lysobacter showing a significant effect of sugar type and concentration on final cell densities (13), we evaluated sucrose, glucose, and maltose each at 1% and 2%. Sugar type and concentration were evaluated by standardizing the protein extract as 1% HiVeg extract. Glucose and maltose at 2% provided the highest levels of Lys-C activity in the culture supernatant. Sucrose at 2% was similar to 1% glucose or maltose. The best medium condition from this test series, which we used in all subsequent 40-L fermentation batches, was 1% HiVeg extract with 2% glucose (including 0.01% each of mono- and dibasic potassium phosphate and 0.02% magnesium sulphate), which yielded 5.6 mg/L Lys-C in culture.
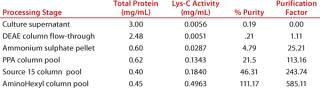
Subjecting crude culture supernatant to a series of four chromatography columns and an ammonium sulphate concentration step leads to an almost 600-fold increase in the Lys-C purity (Figure 2A and Table 1) and gives a single protein band by SDS-PAGE (Figure 2B) when detected by Coomassie blue staining. The purified Lys-C was stable for at least three months (data not shown) when frozen at −20 °C and could withstand at least 10 freeze–thaw cycles (data not shown) without significant loss in activity. We confirmed the pH range for this enzyme to be broad (pH 6–11) with an optimum of pH 9 (Figure 3, TOP).

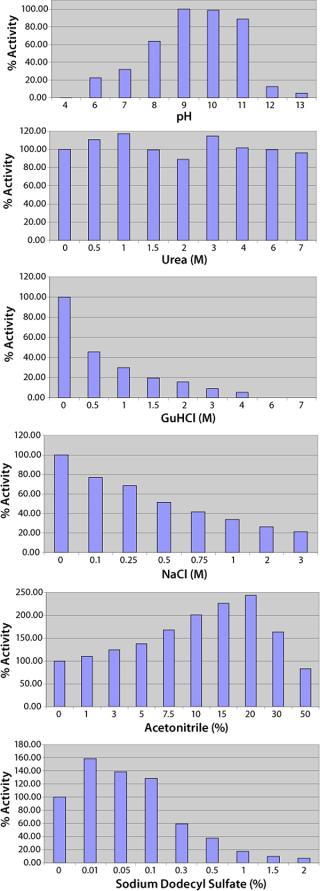
Table 1: Relative percent purity and purification factor for each estp in the Lys-Crecovery process
Consistent with previous observations, Lys-C is not significantly affected by urea up to a concentration of 7 M and SDS to 0.1% (Figure 3). Guanidine hydrochloride and NaCl both have an inhibitory affect on Lys-C, most likely acting through disrupting the enzyme’s electrostatic recognition of the lysine residue in the substrate. Reagents such as acetonitrile that promote electrostatic interactions have the opposite effect, enhancing Lys-C activity, but only up to the point at which the organic component starts to perturb the enzyme (Figure 3).
The linear Lineweaver-Burke plot for change in reaction rate with changing substrate concentration yields values of 78.125 ΔOD405 nm/min and 55 µM for Vmax and Km, respectively (Figure 4A). The serine protease inhibitor Nα-ptosyl-L-lysine chloromethyl ketone (TLCK) is a noncompetitive inhibitor of Lys-C, decreasing Vmax but having little effect on Km (Figure 4B). Other noncompetitive inhibitors were phenylmethyl sulfonyl fluoride (PMSF), 4-(2-aminoethyl)benzene sulfonyl fluoride (AEBSF), and aprotinin, consistent with Lys-C being a trypsin-like serine protease. We saw competitive inhibition with phosphate buffers (Figure 4C), the Ki being 5.3 mM. Other salts (NaCl and KCl) acted as inhibitors. We found that a number of other serine protease inhibitors did not inhibit Lys-C, including N-tosyl-L-phenylalanine chloro-methyl ketone (TPCK) and N-acetyl-L-leucyl-L-leucyl-L-argininal (Leupeptin).
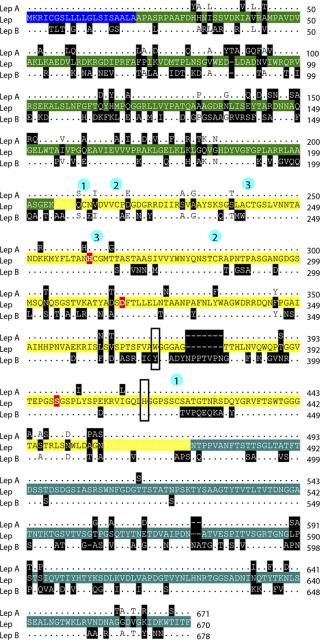
Additional characterization of the Lys-C protein involved cloning the gene encoding it to determine its complete amino-acid sequence. This approach also provided the prepro sequence and C-terminal extension that are absent from the mature enzyme in culture supernatant. We subjected a sample of our purified Lys-C protein to N-terminal sequencing, which yielded GVSGQCNVDVVCP, showing 11 of 13 residues identical to published Lys-C proteins (9, 10).
Using that definitive sequence, together with identified regions of conservation between published Lysobacter Lys-C gene sequences (lepA and lepB), we designed PCR primers to determine the full gene sequence using chromosome walking. The Lys-C gene sequence from Lysobacter enzymogenes (ATCC 27796) is 2,010 bp encoding a 70.5-kDa protein of 670 amino acids. The prepro protein includes a 20–amino-acid secretion signal sequence at its N-terminus, a 184–amino-acid prosequence, a 268–amino-acid mature protease domain, and a 198–amino-acid C-extension prosequence (Figure 5). The catalytic triad in the active site (His57 Asp113 Ser194) is conserved, as is the position of the six cysteine residues (Cys6, Cys12, Cys36, Cys58, Cys80, Cys216) that form three disulfide bonds in the mature protease (14).
Through optimization of the nitrogen source, together with the sugar type and concentration, we obtained an animal-free medium composition suitable for production of Lys-C protease from Lysobacter enzymogenes using a combination of 1% plant-derived protein extract and 2% glucose. Under sugar-limited conditions (at 1% sucrose), yeast and Lupin extracts provided a two- to fourfold increase in Lys-C production over other plant extracts. However, with 2% glucose, the improvement was as much as sevenfold, even when combined with the HiVeg plant extract that had performed poorly with 1% sucrose. Further optimization may be possible through refinement of the plant extract concentration and the KH2PO4 or MgSO4 levels.
The Cellexus Cellmaker Lite 2 fermentor provided a simple, self-contained system for fermentation up to 50-L capacity. The disposable bag technology eliminated a need for resterilization capabilities and simplified maintenance of the system. The volume of the fermentor was also manageable with laboratory-scale centrifugation (using a 6-L capacity rotor) for culture clarification.
Use of sequential column loading dramatically reduces the purification time for proteins and limits conditions for enzyme self digestion. This is of particular interest with proteins that exhibit limited stability or intrinsic proteolytic activity. Endoprotease Lys-C possesses a number of unique attributes that favor this type of purification approach. It retains protease activity in 8-M urea and 20% acetonitrile, which can be useful during its purification. Inhibition of protease activity by high salt concentrations also helps to reduce self-digestion during (NH4)2SO4 precipitation steps and salt elution from an AminoHexyl Sepharose column.
Use of mixed-modal chromatography permitted sample loading at high ionic strength, which is ideal following (NH4)2SO4 precipitation. Removal of sample handling between process operations (dialysis, diafiltration, and ultrafiltration) streamlines the entire process dramatically, reducing overall processing times. Elution with an 8-M urea buffer in a batch mode also served to inhibit contaminating proteases reducing proteolysis. This batch elution proved to be more efficient than gradient elution, which did not provide additional purification advantage and took significantly longer to perform. The eluted 8-M urea sample was directly loaded onto a neutral-pH reverse-phase chromatography column, which provided a degree of purification and buffer exchange. Again the property of Lys-C that allowed it to retain functionality in acetonitrile was crucial to being able to use this step in purification.
The final polishing step, using Amino-Hexyl Sepharose, gave a final purification needed to yield pure Lys-C and could be eluted in a buffer that was suitable for frozen storage. During the entirety of this process, the only manipulation of the sample required between loading columns was a twofold dilution following the reverse-phase step to reduce the acetonitrile concentration, maintaining integrity of the protease and also preventing damage to the Amino-Hexyl Sepharose column.
Our entire purification scheme provided almost 600-fold enrichment of Lys-C in under eight hours of processing time. The order of the steps was essential to the overall process efficiency. Although precipitation of Lys-C directly from the culture supernatant was possible, the increased ionic strength would have limited the usefulness of the anion-exchange step. The high viscosity of the resuspended (NH4)2SO4 pellet caused problems with all but the mixed modal (PPA) chromatography media. Finally, the 8-M urea elution buffer from the mixed-mode column could be purified on reverse-phase media alone because it did not bind to any of the other columns. Hence, we defined the order of the steps in this purification scheme:
-
DEAE
-
(NH4)2SO4 precipitation
-
PPA Hypercel chromatography
-
Resource 15 reverse-phase chromatography
-
AminoHexyl Sepharose chromatography.
Constructing a purification scheme with limited or no manipulation between chromatography steps reduced cost both in terms of time and capital equipment, which makes this process more suitable for larger-scale production.
Comparing the purification scheme maintained at 4 °C with the same scheme at room temperature (22 °C) showed only marginal increase in proteolysis of the Lys-C purified product. This clipping had little to no effect on the specific activity of the resulting purified enzyme. Lys-C self-digestion was often seen with prolonged digestion reactions. However, specific lysine cleavage activity was maintained for over two weeks when the sample was stored at room temperature, indicating that cleavage within the exposed regions of the enzyme does not lower its proteolytic activity.
Enzyme characterization confirmed a broad pH range with an optimum near pH 9.0 (Figure 4), a Michaelis constant (Km) of 55 µM for the Bz-Lys-pNA substrate (Figure 4) which is similar to that quoted for the enzyme derived from Acromobacter lyticus, Km = 70 µM (4), and inhibition characteristics consistent with Lys-C being a trypsin-like serine protease. Serine protease inhibitors that act through arginine residues (e.g., Leupeptin) or act on chymotrypsin-like serine proteases are not inhibitory. The Lys-C activity is negatively affected by reagents that interfere with electrostatic interactions (salts, phosphate) and positively by those that enhance such interactions (organic buffers such as acetonitrile) at low concentrations (Figure 3). As far as we have been able to determine, the proteolytic activity of this new form of Lys-C is identical to the other isoforms that have been studied.
The complete DNA sequence for our secreted lysyl endopeptidase from Lysobacter enzymogenes (ATCC 27796) identified a new isoform of Lys-C. The isolated DNA sequence was verified as encoding the purified protein through a matching N-terminal amino acid sequence GVSGQCNVDVVCP present in the mature protease. Our deduced sequence of the 670–amino-acid Lys-C prepro protein has 87% identity to lepA and 69% identity to lepB, two previously identified Lys-C sequences in Lysobacter. The catalytic triad His57 Asp113 Ser194 is present and is consistent with proteolytic activity. The aromatic residue at position 169, involved in the broad pH optimum for this enzyme (15), is a tryptophan in Lep A and this Lys-C and a tyrosine in Lep B. Although these comparisons suggest this Lys-C is more like Lep A, there are both Lep A- and B-specific deletions/insertions present in the new sequence. In addition, of the 22 amino acids unique to this Lys-C, two changes involve a proline residue, suggesting a possible earlier divergence of all three isoforms.