
STORYBLOCKS (WWW.STORYBLOCKS.COM)
Host-cell proteins (HCPs) constitute a significant class of process-related impurities during biologics manufacturing. Due to their potential impact on product quality and efficacy as well as patient safety, the total amount of residual HCP in a biological drug substance generally is considered a critical quality attribute (CQA) that usually needs to be tested for during batch release (1, 2). It is both an “industrywide” common understanding and a regulatory requirement to remove HCPs from biologics to acceptably low levels that will not affect product quality, compromise expected product efficacy, or jeopardize patient safety (3, 4). However, the complexity and diversity of residual HCP composition in drug substances and limited knowledge publicly available on the biological, physiological, pharmacological, and toxicological effect of individual HCPs detected in biologics create significant challenges. The main difficulty is in effectively determining at what level — and more important, for which specific HCPs — they could be considered safe or low risk. The industry applies HCP specification limits based on experience, process capabilities, clinical development stages, and HCP testing results from sensitive immunoassays supplemented with HCP characterization by orthogonal methods (5–7). However, little to no guidance is available on how to manage HCP-related risks throughout drug substance development.
Certain risks can be controlled during cell line and upstream process development if they are known. For example, lipoprotein lipase (a difficult-to-remove HCP) was knocked out during cell line development in one case to reduce its potential impact on product formulation stability (8). However, genetic engineering at the cell line level can influence the expression of other HCPs inadvertently and thus create new risks. For example, a single-gene knockout in Escherichia coli (E. coli) has shown significant impact on the host cells’ metabolic modes through regulating multiple metabolite concentrations and enzyme activities (9).
HCP-related risks most often are controlled by robust downstream processes. Traditionally, most HCP impurities are neither identified nor characterized, so best efforts are required to remove them through downstream processing to minimize their effects on product quality, safety, and efficacy. During biomanufacturing, downstream processing often balances product yield, purity, throughput, and robustness to reduce manufacturing cost without sacrificing product quality.
Downstream processes often are optimized based on critical process parameters (CPPs) and CQAs. Optimization efforts heavily rely on analytical capabilities to guide decision-makers in determining whether process changes are necessary. Several review articles and the recently published USP monograph chapter 1132 describe in detail the analytical tools available for HCP measurement along with each method’s advantages and limitations (1, 5, 10, 11). Recent developments in massspectrometry–based techniques benefiting from one- and two-dimensional (1D, 2D) separation modes have enhanced downstream process development activities to remove high-risk HCPs (12–14).
Without clear guidance on the acceptable levels of HCP impurities in drug substances, downstream processing groups often are challenged to determine how much removal of HCP actually is necessary. Because of the complexity of this matter, no simple answers exist to questions regarding when HCP removal is sufficient. Rather, that must be decided case by case through science- and risk-based evaluations. For scientific evaluations, HCP identification and quantitation with methods orthogonal to immunoassays (e.g., an LC–MS approach) often are needed to overcome the limitations of HCP enzyme-linked immunosorbent assay (ELISA) methods (5, 15–18). Although analytical characterization of HCP with orthogonal methods can help identify individual HCPs and build up an understanding of their potential impacts, the industry still needs risk assessment and management to guide process development teams in evaluating potential risks and making appropriate decisions based on the level of risk they identify.
In different phases of development, the information available to help evaluating risks is different, so the level of risk identified may not be the same. Often at an early phase of development, only total residual HCP is measured, and the ppm level determined by immunoassay would be used for risk assessment (with levels at ≤100 ppm generally considered to be low risk). Exceptions have been seen when the residual HCPs are mainly made up of a few highly immunogenic or highly active proteins, then the risk has to be assessed accordingly (19, 21).
One example is hamster phospholipase B-like 2 (PLBL2) protein, which can be found in some monoclonal antibodies (MAbs) derived from Chinese hamster ovary (CHO) cell lines. It elicited an immune response in patients treated with lebrikizumab at levels as low as 0.2–0.4 ppm, although the observed response showed no direct link with any clinical adverse effects (19). Another example is group XV lysosomal phospholipase A2 isomer X1 (LPLA2), a CHO HCP with lipase activity that could promote degradation of polysorbate 80 at levels <1 ppm (21).
In regard to addressing HCP-related safety risk case by case, one publication from Genentech (a member of the Roche group) described a risk-assessment framework that takes into consideration multiple factors: HCP identity, host, homology with human counterparts, disease indication, dose and dose duration, patient age and sex, and so on (7). This risk assessment tool was designed mainly for application to clinical safety risks associated with individual HCPs identified in drug products.
Risk control for process development — from preclinical phase to commercialization — often is achieved through process optimization guided by analytical characterization using phase-appropriate assays. During downstream process development, a process-specific or platform-specific HCP assay (commonly a sandwich ELISA) needs to be developed, qualified, and validated for HCP measurement for drug substance (DS) release and in-process testing. Often in the early phases of process development, platform assays and commercially available generic assays are used for HCP measurement and control. They are good tools for HCP clearance studies or process consistency indication but often are limited in HCP coverage because of potentially missing anti-HCP antibodies to process-specific HCPs or nonimmunogenic HCPs (5). Therefore, HCP risk control relies on both robust downstream processing and sensitive analytical methods to guide downstream process optimization.
Bracewell and colleagues (6) have published a holistic review of risk-based HCP control management based on the severity, detection, and abundance of individual HCPs as they are identified. Some other reports also demonstrate that the product-degradation risk of cathepsin D (a CHO host-cell protease that has contributed to degradation of recombinant proteins during early process development) is well controlled and mitigated after process optimization (22, 23). For cases in which trace levels of HCPs still pose product or safety risk even after mitigation efforts, the reassessment of risk would be necessary. For example, PLBL2 triggered a dose-dependent immune response in treated patients with asthma during phase 3 studies even after the level of that protein was reduced significantly through manufacturing process optimization (19).
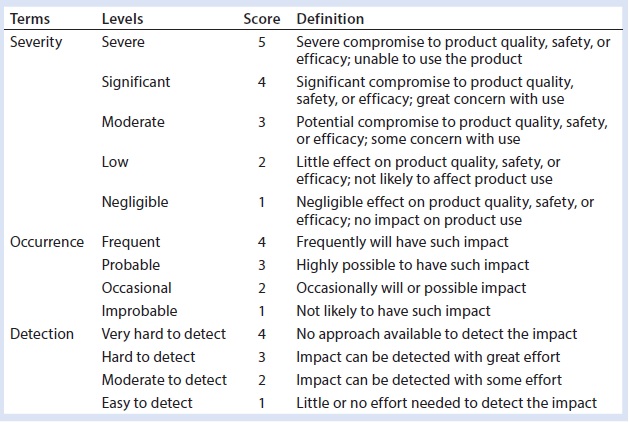
Table 1: Definition of risk assessment terms used herein
To date, however, even with increased understanding of HCPs and their potential risks, no practical approach has been made available for risk management during bioprocess development. Through our collaboration, the BioPhorum Development Group (BPDG) team has identified common HCP-related risk factors and built a template for semiquantitative risk assessment during process development based on publicly available information. To this end, the BPDG HCP working team’s assay and knowledge-sharing experts are focused on establishing a common HCP risk assessment tool and mitigation strategy to guide bioprocess development.
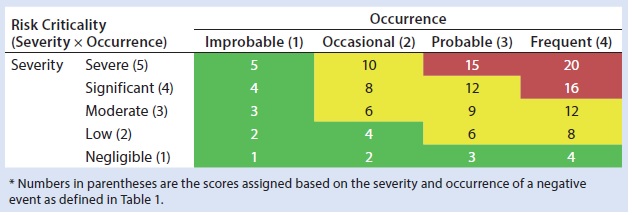
Table 2: HCP risk classification based on risk criticality
Risk Assessment and Management Methodology
An international harmonization guideline emphasizes the use of tools for quality risk management that can enable effective and consistent risk-based decisions made by both industry and regulatory agencies (23). Recent articles on HCP risk assessment have discussed factors that need to be considered and a general framework for performing these risk assessments (6, 7). Here and in Part 2 next month, we discuss a process development HCP risk-assessment tool that can be used for biologics in all stages of development.
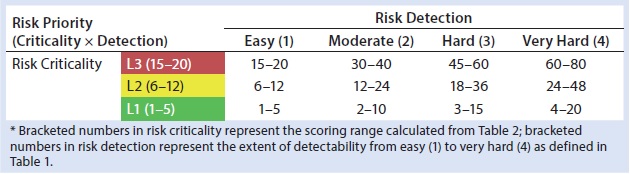
Table 3: HCP risk-priority number (RPN)
Our risk-assessment methodology is based on the failure mode, effects and criticality analysis (FMECA) approach used to identify and rank different potential failure modes and develop appropriate preventive actions to mitigate risks. Our methodology begins with classification of identified risks into three different levels based on their criticality scores, which are derived from multiplying severity (on a scale of 1 to 5, as defined in Table 1) and occurrence (on a scale of 1 to 4, as defined in Table 1) of an HCP-related negative event (Table 2). A risk priority number (RPN) is obtained by multiplying the risk criticality score with a risk detection score (on a scale of 1 to 4, as defined in Table 1) to help prioritize the identified risk (Table 3). Risk priority is classified into three different levels based on the scoring range defined in Table 3.

Table 4: HCP risk-priority definitions and mitigation activities based on the RPN
Table 4 proposes definitions for each risk-priority level and its corresponding mitigation activities. RPNs falling within the high range (≥45) require significant process changes and/or assay improvements for adequate mitigation of potential negative events and to minimize process risks on HCP monitoring and clearance. For medium RPNs (21–44) derived from a risk assessment, either process or assay improvements may be needed to control them. Low RPNs (1–20) derived from a risk assessment require no process or assay improvements. Upon mitigation, risk priorities can be reevaluated until mitigation activities bring them down to a low RPN (≤20). In certain cases, high levels of risk criticality alone (L3) can trigger mitigation activity even if the final RPN is low (≤20). For potential negative events with low RPNs, generally no mitigation activities will be implemented. A formal decision can be made to accept such risks until further analytical characterization and clinical studies identify new ones.
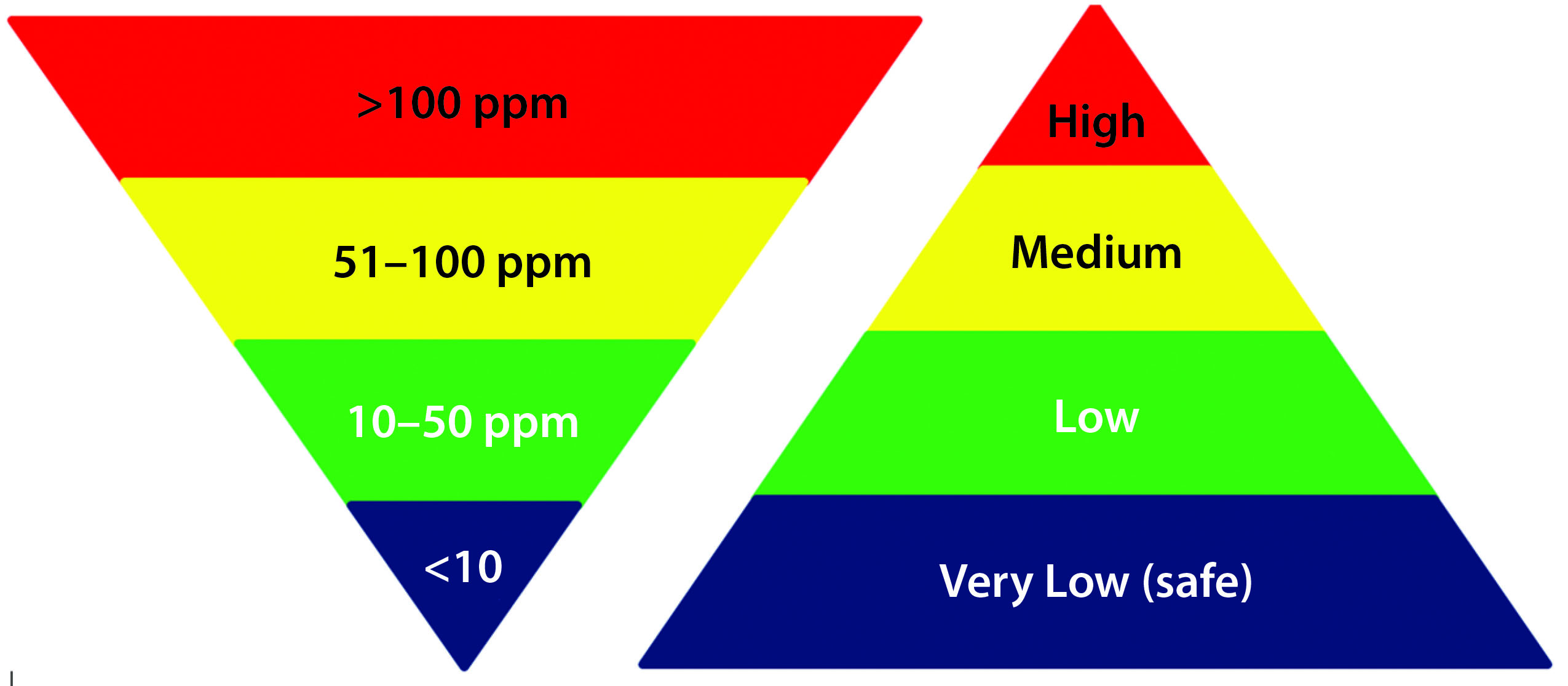
Figure 1: Commonly acceptable risk classification based on the residual HCP amount in MAb drug products
One challenge associated with risk assessment is to align the understanding of FMECA terms to reduce the subjectivity of risk scoring. A reasonable assessment of risk priority often requires terms to be well defined, and team members performing this task must have a common understanding of those terms. In industry practice, crossfunctional teams often include analytical and process members who each may take either conservative or aggressive approaches. Each approach has its own set of advantages and disadvantages. In certain situations, however, conflict of opinions and subjectivity regarding risk extent can inhibit effective decision making. Our risk-assessment tool should aid in minimizing those conflicts and subjectivity involved in risk decisions. It uses a semiquantitative approach for assessing risk criticality and priority, calculating a cumulative RPN from each individual team member’s score and comments. Those inputs allow for a relatively comprehensive evaluation to help a team reach consensus on risk priority and mitigation strategies.
Table 5: TP-A early process development host-cell protein risk assessment by one subject-matter expert (SME)
Case Study: Table 5 presents an example of this risk assessment tool applied in a real-life situation. A platform two-column purification process designed for MAb purification was applied for preclinical development of therapeutic protein A (TP-A). Based on prior experience, the HCP specification limit for this early phase program DS is set at <100 ppm. A generic commercial ELISA kit was used to measure residual HCP levels in DS and process intermediates. Assay results showed >100 ppm of residual HCP levels in TP-A. Although the composition and impact of HCPs were not identified in this situation, the negative event of residual HCP levels exceeding the platform specification could compromise product quality, safety, and/or efficacy.
Consequently, the severity score for that negative event is defined as 5 by a subject matter expert (SME) on HCP analysis. The potential for residual HCP at 100–200 ppm (detected range in DS) to cause significant product quality or safety concerns is likely, with an occurrence defined at 3 by the same SME, leading to a risk-criticality score of 5 × 3 = 15 (Table 5). At this early stage of development, with limited analytical characterization information available, the detectability of potential HCP risks at 100– 200 ppm level is considered to be very difficult. Thus a score of 4 was assigned by the same SME according to Table 1 and 2, leading to a final RPN of 5 × 3 × 4 = 60, which falls within the high-risk priority range. In this situation, high risk should be mitigated with process changes to reduce total HCP amounts further and thus minimize the potential impact from high levels of residual HCPs in a DS (Table 5).

Table 6: TP-A early process development host-cell protein risk assessment by the chemistry, manufacturing, and controls (CMC) team
As mentioned above, a major challenge in performing risk assessments is the degree of subjectivity involved in defining the severity, occurrence, and detection of impact for a given negative event. To address that concern in this particular case, four team members from the chemistry, manufacturing, and controls (CMC) team were selected to perform the risk assessment: two from the analytical group and two from the process group. Those team members had slightly different views on severity, occurrence, and detection (Table 6), but the final risk-priority score from that four-person team averaged out to be in the same high risk-priority range as the initial evaluation in Table 5.
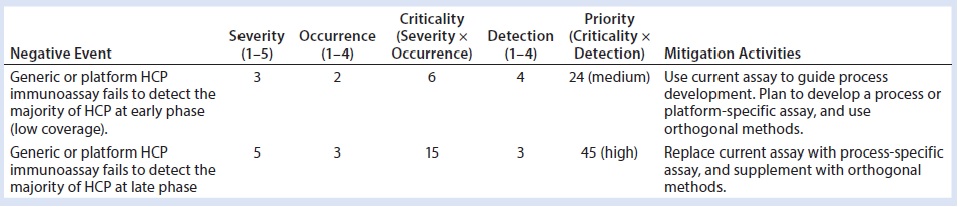
Table 7: Risk assessment on the use of generic host-cell protein immunoassay for early and late phases in development
Potential Negative Events
Members of the BPDG HCP working team have compiled a list of potential negative events related to HCPs during process development. This list was generated mainly from experience and published studies, and it is divided into two categories of risks encountered during early and late-phase process development: process and analytics. That classification should help team members performing risk assessments to be more specific about impacts from different causes of negative events (process or analytics) and corresponding mitigation strategies to reduce those risks according to the development phase where they occur. For example, a generic HCP immunoassay that fails to detect most HCPs poses relatively lower risk at an early phase of development than in late-phase development because the severity, occurrence, and detection all vary depending on the phase of development (Table 7).
Below and in Part 2, we describe some HCP-related negative events occurring during bioprocess development, along with examples of how the risk assessment tool can be applied for each type of event. A more comprehensive list of such events, with risk assessment and corresponding mitigation strategies, was created by the BPDG’s HCP working team in a spreadsheet that includes details on the cause, effects, and mitigation activities for each negative event. These are commonly observed incidents across the biopharmaceutical industry, so the risk assessment provides guidance on how to control HCP-related risks tied to such negative events.
High Levels of Residual HCPs in Drug Substance: Often in the early phase of process development, HCP levels are monitored using a platform HCP immunoassay or commercially available ELISA kit. HCP levels are generally expected to be controlled at <100 ppm for biologics, especially MAb products that go through affinity purification (10). On some occasions, however, residual HCP levels are measured as >100 ppm in DS — often because of the unique properties of a new product — requiring further optimization of its platform purification process to remove HCPs to acceptable levels. For example, when a product’s isoelectric point (pI) is similar to that of most HCPs present, it is difficult for charge-based purification procedures to remove HCPs effectively, so further process improvement would be required.
Table 5 is one example of risk assessment for high residual HCP amount in DS. When residual HCP levels of >100 ppm are measured in DS with a qualified method, it is relatively straightforward to conclude unanimously that process changes should be considered for mitigating the risks associated with such high levels of HCP. However, when HCP levels fall within the range of 1–100 ppm, although the associated risks can be classified generally as in Figure 1, the situation is not as clearcut. The impact of HCP on product quality, safety, and efficacy not only depend on the total amount but also the composition and amount of individual HCP (19, 20, 22, 24–26). The risk-assessment template described herein overcomes those difficulties by taking into account the potential impact (severity) of an event, its probability of effect (occurrence), and detectability of potential effects (detection) at three dimensions to define both risk priority and mitigation. Often a risk assessment using those three dimensions yields a relatively objective evaluation of the potential risks during process development before locking down a biomanufacturing process for phase 3 and commercial production.
Looking Ahead
Next month in part 2, we will explore more potential negative events related to HCPs during bioprocess development — dilutional nonlinearity issues with sample testing and products with unusual stability profiles and/or degradation pathways — as well as other factors to consider when assessing HCP-related risk. We will provide a real-world industry case study of a heat-shock protein. And we will further consider other applications of the risk-assessment tool as well as its limitations.
References
1 Wang X, Hunter AK, Mozier NM. Host Cell Proteins in Biologics Development: Identification, Quantitation and Risk Assessment. Biotechnol. Bioeng. 103(3) 2009: 446–458; doi:10.1002/bit.22304.
2 Jin M, et al. Profiling of Host Cell Proteins By Two-Dimensional Difference Gel Electrophoresis (2D-DIGE): Implications for Downstream Process Development. Biotechnol. Bioeng. 105(2) 2010: 306–316; doi:10.1002/bit.22532.
3 Points to Consider in the Manufacture and Testing of Monoclonal Products for Human Use. US Food and Drug Administration: Rockville, MD, 1997.
4 CPMP/BWP/382/97. DNA and Host Cell Protein Impurities, Routine Testing Versus Validation Studies. The European Agency for the Evaluation of Medicinal Products, Human Medicines Evaluation Unit: London, UK, 6 October 1997.
5 Zhu-Shimoni J, et al. Host Cell Protein Testing By ELISAs and the Use of Orthogonal Methods. Biotechnol. Bioeng. 111(12) 2014: 2367–2379; doi:10.1002/bit.25327.
6 Bracewell DG, Francis R, Smales CM. The Future of Host Cell Protein (HCP) Identification During Process Development and Manufacturing Linked to a Risk-Based Management for Their Control. Biotechnol. Bioeng. 112(9) 2015: 1727–1737; doi:10.1002/bit.25628.
7 de Zafra CL, et al. Host Cell Proteins in Biotechnology-Derived Products: A Risk Assessment Framework. Biotechnol. Bioeng. 112(11) 2015: 2284–2291; doi:10.1002/bit.25647.
8 Chiu J, et al. Knockout of a Difficult-to-Remove CHO Host Cell Protein, Lipoprotein Lipase, for Improved Polysorbate Stability in Monoclonal Antibody Formulations. Biotechnol. Bioeng. 114(5) 2017: 1006–1015; doi:10.1002/bit.26237.
9 Zhu J, Shimizu K. Effect of a Single-Gene Knockout on the Metabolic Regulation in Escherichia coli for d-Lactate Production Under Microaerobic Conditions. Metab. Eng. 7(2) 2005: 104–115; doi:10.1016/j.ymben.2004.10.004.
10 Eaton LC. Host Cell Contaminant Protein Assay Development for Recombinant Biopharmaceuticals. J. Chromatogr. A 705(1) 1995: 105–114.
11 Hogwood CE, Bracewell DG, Smales CM. Measurement and Control of Host Cell Proteins (HCPs) in CHO Cell Bioprocesses. Curr. Opin. Biotechnol. 30, December 2014: 153–160; doi:10.1016/j.copbio.2014.06.017.
12 Kreimer S, et al. Host Cell Protein Profiling By Targeted and Untargeted Analysis of Data Independent Acquisition Mass Spectrometry Data with Parallel Reaction Monitoring Verification. Anal. Chem. 89(10) 2017: 5294–5302; doi:10.1021/acs. analchem.6b04892.
13 Doneanu CE, et al. Enhanced Detection of Low-Abundance Host Cell Protein Impurities in High-Purity Monoclonal Antibodies Down to 1 ppm Using Ion Mobility Mass Spectrometry Coupled with Multidimensional Liquid Chromatography. Anal. Chem. 87(20) 2015: 10283–10291; doi:10.1021/acs.analchem.5b02103.
14 Doneanu CE, et al. Analysis of Host-Cell Proteins in Biotherapeutic Proteins By Comprehensive Online Two-Dimensional Liquid Chromatography/Mass Spectrometry. MAbs 4(1) 2012: 24–44; doi:10.4161/mabs.4.1.18748.
15 Valente KN, Lenhoff AM, Lee KH. Expression of Difficult-to-Remove Host Cell Protein Impurities During Extended Chinese Hamster Ovary Cell Culture and Their Impact on Continuous Bioprocessing. Biotechnol. Bioeng. 112(6) 2015: 1232–1242; doi:10.1002/bit.25515.
16 Thompson JH, et al. Improved Detection of Host Cell Proteins (HCPs) in a Mammalian Cell-Derived Antibody Drug Using Liquid Chromatography/Mass Spectrometry in Conjunction with an HCPEnrichment Strategy. Rapid Commun. Mass Spectrom. 28(8) 2014: 855–860; doi:10.1002/rcm.6854.
17 Zhang Q, et al. Comprehensive Tracking of Host Cell Proteins During Monoclonal Antibody Purifications Using Mass Spectrometry. MAbs 6(3) 2014: 659–670; doi:10.4161/mabs.28120.
18 Ahluwalia D, et al. Identification of a Host Cell Protein Impurity in Therapeutic Protein, P1. J. Pharm. Biomed. Anal. 141, July 2017: 32–38; doi:10.1016/j.jpba.2017.03.065.
19 Fischer SK, et al. Specific Immune Response to Phospholipase B-Like 2 Protein, a Host Cell Impurity in Lebrikizumab Clinical Material. AAPS J. 19(1) 2017: 254–263; doi:10.1208/s12248-016-9998-7.
20 Hall T, et al. Polysorbates 20 and 80 Degradation By Group XV Lysosomal Phospholipase A2 Isomer X1 in Monoclonal Antibody Formulations. J. Pharm. Sci. 105(5) 2016: 1633–1642; doi:10.1016/j.xphs.2016.02.022.
21 Bee JS, et al. Trace Levels of the CHO Host Cell Protease Cathepsin D Caused Particle Formation in a Monoclonal Antibody Product. Biotechnol. Progr. 31(5) 2015: 1360– 1369; doi:10.1002/btpr.2150.
22 Robert F, et al. Degradation of an Fc-Fusion Recombinant Protein By Host Cell Proteases: Identification of a CHO Cathepsin D Protease. Biotechnol. Bioeng. 104(6) 2009: 1132–1141; doi:10.1002/bit.22494.
23 ICH Q9: Quality Risk Management. US Fed. Reg. 71(106) 2006: 32105–32106; www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q9/Step4/Q9_Guideline.pdf.
24 Dixit N, et al. Residual Host Cell Protein Promotes Polysorbate 20 Degradation in a Sulfatase Drug Product Leading to Free Fatty Acid Particles. J. Pharm. Sci. 105(5) 2016: 1657–1666; doi:10.1016/j.xphs.2016.02.029.
25 Jawa V, et al. Evaluating Immunogenicity Risk Due to Host Cell Protein Impurities in Antibody-Based Biotherapeutics. AAPS J. 18(6) 2016: 1439–1452; doi:10.1208/s12248-016-9948-4.
26 Mihara K, et al. Host Cell Proteins: The Hidden Side of Biosimilarity Assessment. J. Pharm. Sci. 104(12) 2015: 3991–3996; doi:10.1002/jps.24642.
Fengqiang Wang, Daisy Richardson, and Hans-Martin Mueller are with Merck Research Laboratories, Merck & Co. Inc. (Kenilworth, NJ). Georgeen Gaza-Bulseco is with AbbVie Bioresearch Center (Worcester, MA). Xinrong Liu is with ImmunoGen, Inc. (Waltham, MA), as was Fang Liu when this work was done. Yiwei Zhao is with Takeda Pharmaceuticals (Cambridge, MA). Satish Sharma is with Bristol-Myers Squibb (Devens, MA). Markus Haindl is with Roche Diagnostics GmbH (Penzberg, Germany). Carl Co is with Biogen (Cambridge, MA). Direct correspondence to Tony Gill (gilla@biophorum.com), BioPhorum Development Group (BPDG) facilitator. Since its inception in 2004, the BioPhorum has become a trusted environment in which senior leaders of the biopharmaceutical industry come together to share and discuss openly the emerging trends and challenges facing their industry. BioPhorum currently comprises more than 2,000 active participants in six “phora” covering drug substance, development, fill–finish, a technology roadmap, information technology, and supply partners. The HCP Collaboration is part of the Development Group. This article is a composite view of opinions shared by the whole of the BPDG-HCP and should not be attributed to the individual positions of the participating companies.