Biopharmaceutical drug products are not only well established but also contribute to a large degree to new drug entity filings. Currently approved biopharmaceuticals and proteins are now widely used to treat diseases as diverse as cancer, autoimmune disorders, myocardial infarction and various growth factor deficiencies. The unmet medical need can be so essential, such as a novel approach to cancer treatment, that biotech companies will choose to defer the optimal design of the production process to reduce the time-to-market. In other instances, the manufacturer may choose to defer investment in large-scale production facilities until a real-world understanding of the market potential of the new product is determined. The cost and start up times for large-scale protein production facilities is substantial, typically ranging from $500 million to $1 billion and taking approximately 3–5 years to achieve full regulatory approval. In addition, the timelines for modifications to existing facilities are also significant and are often of the order of 1–2 years from initiation to final approval by a major regulatory agency. As a result, it is not uncommon for a manufacturer to launch a new product with a yield and/or production process capacity or output that is suboptimal. Once the product reaches the market, the additional burden will be to cover the drug product demand as fast as possible. Given the large investment in time and cost for new facilities, some drug developers build large inventories to compensate before the product is launched into the market, collaborate with contract manufacturers or even partner with competing companies. All of these generally add significant risk and cost, which is certainly not optimal from the viewpoint of the accounting department of the drug developer.
Additionally, the potential cost pressures of biosimiliar or generic biotechnology products is driving the biotech industry to seek increased efficiencies by revising existing processes and evaluating new technologies. Former FDA Commissioner, Dr Mark B. McClellan, confirmed before the joint economic committee of the US congress (2003) that not only does GMP regulation require revision to fit the progress of manufacturing technologies but new technology advancements need to be encouraged to be able to reduce development hurdles and generate more affordable drug products. He also made it clear that generics will play a greater role and cover a larger patient base in the future. This said, the industry understands the underlying, increasing competitive pressure and is adjusting to this challenge by the introduction of state-of-the-art process designs. This paper will address the pro and cons of existing processes and will venture into the potential future enhancements of biopharmaceutical processing that may be realized in the next decade.
Biopharmaceutical Processes: Past and Present
Current biopharmaceutical production processes consistently produce proteins that exceed purities of 99.99% or greater and are highly homogenous with respect to size or charge variants. These levels of purity and reproducibility are essential, given the importance of correct three-dimensional conformation for potency and the potential for impurities or product variants to elicit an immune response in the patient. These high quality standards result in drug products that help a multitude of patients with formerly untreatable ailments (with few side-effects that are attributable to product variants or impurities). As the assessment of three-dimensional protein conformation is still an evolving science, process and product consistency and stability should not be taken for granted. Rigorous process development and technology transfer within the biologics industry is essential to stabilize the production process and apply proper process controls to ensure reproducibility. In some instances, though, early clinical success has driven processes to be scaled up to process scale utilizing the early clinical stage equipment or facilities. In addition, the first generation biotechnology products and their production processes are now approximately 20 years post-approval. Many of these products still address significant medical needs but suffer from ageing process technology that often lacks competitive yields and transferability. These situations can involve significant risk to post-approval scale and site transfers given the highly process- and equipment-dependent nature of biotechnology products.
Such instances of rapid clinical success and early stage technology have led to the use of ineffective, non-optimal equipment on the upstream side; for example, thousands of roller bottles instead of a stirred tank reactor. Furthermore, most biotechnology processes started with low cell expression levels of the desired product and concentrations in the 10–50 mg/L area. To obtain the required grams of drug target, large volume fermentation systems or, as stated, thousands of roller bottles found their use. The downstream processes, which follow, were required to handle large volumes, which resulted in large system sizes, often with respective hold-up volumes. Even before the concentrated protein solution reached the purification stage, high yield losses were observed and found its pinnacle in the chromatographic stages of the process. In some instances, target protein losses of up to 70% from the upstream to the final formulation step have been observed. This meant that the ineffective capacity utilization of these bioprocesses resulted in elevated cost of goods sold, which again has an effect on the patient and the affordability of the drug product.
In the last 10 years, biotechnology drug products have become widely effective and established; the demand for monoclonal antibodies, for example, has risen from kilogram levels to the hundreds of kilograms. This meant that cell culture researchers were requested to find either new expression systems or rates. Cell productivity moved from tens of milligrams per litre to a double-digit gram level. Fermenter volumes of 4000 L could achieve the same protein output that formerly required 20,000 L (Figure 1). This cellular output, though, had an immediate effect on the downstream process, which now had to handle the elevated protein concentration, not to mention the cell debris and host cell contaminants. Cell harvest steps were not confronted with the large volumes required in the past, but with a far higher cell density. As the harvest step utilized either centrifugation, depth or cross-flow filtration, systems had to be optimized to avoid premature fouling and yield losses due to hold-up volume or capture within the harvest step. Often, harvest step optimization could be accomplished, but the resulting higher protein concentration caused new challenges in the next step — purification.

Vince Anicetti VP Quality Genetech

As the yields of cell culture processes have improved (2–4 g/L is not uncommon for new monoclonal antibody processes), the bottleneck moved from the upstream side to the downstream process, especially the chromatographic steps, as the equipment has a dimensional limitation. The largest affinity chromatography column experienced has a diameter of 3 m, which is difficult enough to handle, especially when packing and cleaning such equipment. Having said this, columns of this size pose a capital expense that often cannot be stomached by smaller biotech organizations. The alternative for these organizations is to move to contract manufacturing organizations. Affinity chromatography has greatly simplified protein recovery processes, particularly in the use of Protein A-coupled resins for monoclonal antibody products. However, the Protein A resin has its own expense, often in the millions of dollars, which means that the columns need to be utilized to an exhaustive extent. Despite the high selectivity and platform-enabling properties of Protein A affinity resins, its price, in $/L of resin or $/g of Mab purified, is an order of magnitude higher than that of conventional non-affinity chromatography media, accounting for up to a third of the overall raw material costs. This is a major challenge to the development of cost-efficient manufacturing processes. Other disadvantages of Protein A include the intolerance of most protein A ligands to the caustic regeneration and sanitization used in conventional chromatography media (1). Furthermore, Protein A released from an affinity column needs to be removed, which means additional, preventive steps must be implemented, such as ion exchange chromatography.
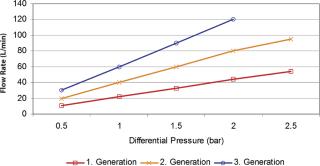
Because the dimension of the column is a limiting factor, the flow through such columns is restricted; the time to process the protein concentrations mentioned will take up time. This causes a “stop and go” effect; the production process pulses instead of running in a steady flow. If harvested product cannot be moved to the next step, some organizations resolved to freeze the bulk material with the effect of additional yield losses and idle production capacity. Traditional columns run slowly, which can have a detrimental effect on some of the targeted proteins. Experience has showed that processing time is of essence to avoid enzymatic breakdown of the proteins and to perform operations at ambient temperatures, which is essential for large-scale production processes. Running traditional chromatography columns also means a large buffer consumption to wash and sanitize the systems. New technologies were asked for to overcome the bottleneck of traditional chromatography speed, like the introduction of membrane chromatography, especially in the ion exchange column area (polishing) (2) (Figure 2). The flow rate through these porous membrane materials is many times (5–10) faster. Binding capacity, in traditional bead systems dependent on diffusive flow into the bead, is available on the surface of the porous structure and of immediate availability, which can result in a 100-fold capacity increase. Buffer flush volumes are significantly lower, which consequently reduces the buffer preparation requirements for water-for-injection use or set-up times. The reduction in buffer use can result in savings of tens of thousand of dollars. Membrane chromatography systems can be used repeatedly or as a disposable. As membrane chromatography does not require packing, but rather has the same installation criteria as commonly used membrane filter cartridges, the set-up time requires just minutes instead of hours. Set-up times within existing processes cause major reductions in capacity utilization, often reducing the batch output. Alternative approaches likely to advance in coming years will be the use of non-affinity recovery processes for monoclonal antibodies. Follman (3) has demonstrated the feasibility of a non-affinity monoclonal antibody recovery process and Low (4) has reported an approach combining precipitation, crystallization, extraction and high performance tangential flow filtration. High performance tangential flow filtration has also been combined with cation ion exchange capture to recover monoclonal antibodies (5).
Because the purification step can cause a major disruption within the biologics process, formulation and filling requires a high degree of flexibility. If such flexibility is unfeasible, organizations resolve to either bulk hold, freeze or utilize a contract filler. Careful fluid handling and stability plays a major role when purified bulk product is stored or shipped. Temperature conditions, overlaying gas or freeze conditions can have an adverse effect on the product. Strategies to handle the purified bulk have to be evaluated in the development phase to avoid any undesirable surprises. Any hold step does not usually contribute to value. An additional consideration within production processes is the utilization of cleanroom area (6). In the past, most of the processes were run within high-grade cleanrooms, in some cases even cleaning and set-up. Footprints within higher class cleanroom areas have high running costs. Newer facility designs utilize a closed systems approach, which means that some of the equipment used does not need to be positioned within classified areas, as the barrier on these systems can be membrane filters or aseptic connection between equipment. In some cases, the facility is used for multiple product lines, which considers appropriate segregation and cleaning cycles. As the number of approved protein pharmaceuticals increases, there is an increasing need to improve the flexibility of production facilities. This is important to accommodate smaller volume products as well as providing the optimal return on investment from production facilities. To avoid such fluctuation or idle production capacity, multi-product lines are desired — even if this means a high degree of validation and proof that cross-contamination will not happen.
Biopharmaceutical Processes: The Future
Future biopharmaceutical processes will be driven by multiple factors:
- Multi-product utilization
- Lack of capital
- Fast track production capacity increases
- Equipment utilization
- Increased recovery process capacity.
Evaluating the individual points, multi-product facilities are highly desirable, but come with the added expense of tedious validation work to ensure cross-contamination is minimized. This escalates significantly as the number of processes and fixed equipment increases. Cleaning cycles of reusable equipment is therefore a major focus and often generally performed at a level that ensures the highest probability of acceptance by any regulatory agency. Thus, realizing this capacity utilization comes with considerable cost and changeover investment. The development of a new drug entity takes years and the failure rate is high. Generally accepted estimates range from 8–10 years to reach approval for a new molecular entity, with a cost of approximately $800 million. Capital expenditure is at a minimum in the early stages, due to the attrition rates of far greater than 50% for typical biotechnology new molecular entity candidates. In some cases, the organization doesn’t achieve the capital to build a multimillion dollar facility or acquire equipment such as bioreactors or chromatography skids. Often, the lack of capital results in partnerships with other organization to produce and distribute the product or contract manufacturer utilization.
For example, the threat of pandemic avian flu showed that rapid production capacity increases are not too far distant, but could become an urgent need in the case of an outbreak. These processes cannot be built within a targeted 3-month period, but require that existing facilities are occupied and rapid, flexible production process scenarios installed to run a cell culture process. As some of the facilities might not be positioned in a controlled cleanroom area, the individual process steps need to be presterilized, closed systems that require aseptic connectivity. Additionally, depending on the cell culture process, the individual, modular process steps can be fit to scale, depending on the fluid volumes or protein concentrations processed. Fast track, flexible build-up of a cell culture and recovery process is, or at least will, be a pressing necessity in the future. As discussed, equipment utilization depends very much on the set-up times required. Examples have shown that set-up, which includes cleaning, assembly, sanitization or sterilization, drying and transport, can take up to 15 hours, depending on the piece of equipment. Even a 100 L hold vessel can take up to 5–8 hours to be fully set up. The cleaning process itself could be slowed down if water-for-injection (WFI) demand occur at peak times and vessel rinses are not possible. The entire process does not add to capacity extension, but shortfall. Besides the loss in capacity utilization, potentially lower batch cycles and cleaning a 100 L vessel can cost up to $10,000, depending on the WFI costs, including the work hours, energy and cleaning agent costs. Even if the costs are lower, it will never reach the far lower cost of a 100 L presterilized disposable bag.
All five factors can be addressed by disposable equipment, either as a hybrid or completely disposable format. Disposables contribute to a number of critical future needs for the production of biopharmaceuticals. They largely eliminate most cleaning requirements, reduce capital investment in facilities and equipment, decrease the risk of cross-contamination and provide faster campaign turnaround times. Disposable equipment like hold or mixing bags, vent or liquid filters or chromatography devices have been introduced and used for the last 10 years. However, the feasibility of these disposable bag systems has increased significantly with the evolution of capacities of greater than 1000 L and modular systems for bag support and transport. Such equipment has a proven track record. New to the equipment portfolio is aseptic connectivity of the equipment, bundling of multiple disposable equipment formats, UF/DF devices and bioreactors.
Hold bags are used mainly in media and buffer preparation. Encapsulated filters are attached to a mixing bag and the mixed fluid transferred through this filter into a hold bag. The filled media or buffer bag can be utilized at the point of use and connected via sterile connections attached to the bag or the bioreactor or column. Bags are also used to store intermediates after cell harvest or individual purification steps. Disposable mixing bags can be used for viral inactivation applications. In some cases, the bags are also used to store purified bulk product. This fact necessitates the need for close controls on the quality and stability of the utilized bag product. Manufacturers of bags have to have their films thoroughly evaluated to comply with multiple qualification specifications, which include at least tensile strength, chemical compatibility, gas permeation, extractables/leachables, plastic class tests in accordance with USP, gamma sterilizability, long-term shelf test, unspecific adsorption and particle release. These evaluations are the basis of end-user process validation protocols and can be a helpful tool to determine the compatibility of the bag material with the filled product and/or processing conditions. Similar tests are performed with all other disposable equipment, whereby the test requirements vary to gain the evidence of equipment performance within the individual process. Viral and sterilizing grade filters, for example, have to undergo challenge tests to verify retentivity and performance under the process condition with the drug product or, if impossible, a placebo.
Disposable bioreactors, which are fairly new to the disposable field, are becoming more and more available with various volume ranges and agitation technologies. For some time, rocking motion disposable bioreactors were the only option and were used mainly as seed reactors or as clinical material reactors. Nevertheless, the industry’s experience has always been with reusable, stainless steel stirred tank systems. Feeling comfortable with that agitation modus, the biopharmaceutical industry welcomed first technology options, which utilized stirred agitation within a disposable reactor. The volumes of these reactors range from 50–2000 L. As the yield of cell culture systems continues to increase, production scales at the 1000–2000 L range are increasingly practical. Cell culture processes for monoclonal antibodies are now approaching 5 g/L and it is not unrealistic to expect these levels to more than double in the next decade. Newer designs on the horizon will bring new agitation possibilities to the choices of process development, which in itself creates flexibility and optimization for individual applications. To run such reactors reliably, disposable sensor technology is being developed in a rapid way. So far, pH and dissolved oxygen sensor technologies have proven favorable. Additional sensor variations are in the pipeline and could potentially be utilized on existing controllers.
To obtain secure connectivity between disposable equipment bundles, such as bag/filter, bioreactor/cell harvest, membrane chromatography/multiple bag set, reliable aseptic connectivity is required. This can either be fulfilled by tube welder or sealing systems, which either close and disconnect a system from another module or connect one module to another. A different connectivity option is created by sterile connectors, which can be attached to each other without impinging on the environment. These unique connections allow fast connections, obviating any other equipment for welding or sealing. The progress of disposable technologies into all areas of bioprocessing, in conjunction with connectivity, might enable end-user processes to become fully disposable or at least hybrid solutions in the near future. To design such processes, engineering and automation know-how has to work hand in hand to handle the various fluid streams with appropriate timing and controls. Process analytical technology tools, used now and enhanced in the future require fitting the disposable prerequisites. Increasingly, in-line sensors will be used for routine tests such as pH and protein concentration. In the next decade, online measurements are likely to extend to endotoxin, bioburden and various measures of protein homogeneity. Control systems and data acquisition can be employed in the same fashion, except that disposable sensor technology needs to be able to connect to such controllers. One additional aspect should not be overlooked; the disposable system would be available to the end-user in a modular design and functionality. These modules require appropriate labelling to ensure the user connects the modules in a right format. RFID (radio frequency identification) tags could be use to identify the system’s design parameters and technical specification as well as to determine that “A” needs to be connected to “B”. RFID technology not only allows data storage, but also traceability and connectivity. An RFID-driven connection would allow the fast build up of a disposable process without the risk of undesirable modular blocks being connected to each other.
More and more often, bulk products are shipped from one location to another. Most of the time, such shipment needs to be performed in a frozen condition. However, freezing a bulk protein solution can cause concentration gradients, which can alter or degrade the protein. Freezing and thawing has to be performed in a controlled manner to avoid such gradients. New disposable technology recently became available, which allows a controlled freezing of a volume that can be easily handled by routine transport pods. The maximum volume of such system lies at around 16 L, which may be too low for the user, but also creates certain risk mitigation in case something happens during transport. New technology requirements are needed in the affinity purification step. Protein A is extremely costly, can be a contaminant in itself and requires large stainless steel systems with a large footprint. The resins are reused many times to gain an appropriate return on investment, which means that cleaning of such columns has to be performed with diligence, but also high running costs. To overcome the disadvantages of Protein A, the industry has started to revitalize former efforts to use crystallization or precipitation as a purification methodology. To be able to purify even higher protein concentrations in the future, such “old” technologies might find new preferences.
Upgrading Existing Processes: Possibilities
Many times, process upgrades are deferred because of the potential difficulty in managing inventory transitions across multiple regulatory territories, due to significant differences in submission requirements and review time lines. This may potentially be the case; however, it is generally a better option than running a process with elevated costs or inefficient utilization, especially for a product under generic pressure considerations. Frequently, process design improvements and scale up investments are a calculated deferral when the probability of clinical success is low. The alternate is to risk valuable future R&D dollars in large-scale or state-of-the-art processes that may lack a product to process. However, the consequence of this strategy is that the process frequency lags with obsolete or inefficient technology once the drug product hits the market. Having said this, it becomes obvious that the biologics industry is realizing that the tremendous value losses within inefficient processes cause an undesirable and uncompetitive elevation of the cost of goods sold. Sooner, rather than later, process optimization and investments into new technologies will become essential to be able to compete when the patent is void. The costs involved in filing a post-approval change might look unreasonable, but when compared with the existing batch-to-batch value losses or market value loss when the patent protection runs out, it shows that process improvements are worthwhile. Additionally, the total regulatory review time can be eased substantially if the production process changes are made during late-stage clinical development and included as part of the clinical trial strategy. A post-approval supplement has to be filed when steps are changed close to the final formulation or the final purified product stream.
The trend toward more flexible production facilities will be driven by the need to produce low volume or low margin products, as well as by increasing cell culture yields. However, media supplies on the upstream side could be considered important, but non-critical. Any hardware system that requires cleaning, sterilization and lengthy set-up could be exchanged with a disposable system, which can be set up in minutes. The speed of set-up increases capacity utilization and reduces cleaning steps and costs. Buffer preparation could receive the same attention to reduce set up and cleaning times. Recent membrane filtration system designs tripled flow rate and double total throughput compared with counterparts that are just 10 years older (Figure 2). A trend within the vaccine industry is to discard cross-flow cassettes and utilize these only once to avoid tedious cleaning and set-up requirements. All these equipment optimization possibilities do not necessarily mean that the drug manufacturer has to start from scratch when it comes to drug stability testing, but could test up-to-date technology and utilize such tests to check comparability. When in doubt, it is best is to contact the appropriate regulatory agency and explain what the goal and what the changes are.
Value creation within a biologics process, though, does not inevitably need to start with equipment upgrades. There are simpler optimization activities that have an immediate impact on capacity utilization or yield gains. The most common oversights in existing processes are excessive hold-up volumes, due to inadequate tube/pipe length, vessel designs, oversized filtration systems or process steps that could be avoided. Hold-up volumes, especially at the purified product end, can amount to millions of dollars of losses. In an industry of low volume/high value products, hold-up volume plays an essential role as a cost driver. As the biotechnology industry matures, cost and flexibility will become increasingly significant factors in product development and in life cycle extension. Decreasing production costs through greater facility utilization and lowering the “development go” return on investment threshold will increase the number of new drug candidates that may help patients in the future. Before any changes are made, optimization potentials have to be determined. As a drug manufacturer is usually an expert in one area or application, but not necessarily in all the technology steps utilized, it is advisable to consult the technology supplier to check what could be done to optimize either the process or a particular unit operation. Some vendors offer process surveys that could, at least, create a thorough check-up of the process and a respective report, which could be used in phases by process engineers within the facility. When improvements are found, such suggestions need to be verified by trial work. Again, the vendor can be used to perform trial work with small-scale devices to verify that there is an optimized step or possible solution. Resources within biopharmaceutical facilities are limited, which can delay or even stall the mindset of change requirements. The use of vendor experts would overcome potential resource limitations and would additionally shorten trial work. Obviously, the drug manufacturer is the application expert and the vendor is the equipment expert. The combination of this expertise will allow optimization. Both parties have to rely on each other to make an optimization step successful.
Conclusion
Numerous biopharmaceutical processes are established and run, to various degrees, either optimally or at least effectively. The demand curve for these drug products is increasing rapidly, which either requires increased production capacities or the optimization of existing processes, which in itself are capacity increases. The latter is often regarded as a high regulatory hurdle, but does not necessarily need to be. It is easier to optimize processes or implement new, more efficient technologies, than building a new facility. Multiple new technology opportunities have been discussed. These technologies do not need to be favorable for some process steps or applications; however, considering or evaluating the technology will do no harm. Most of the time, surprising advantages can be found. The disposability of certain process steps in a hybrid scenario or even totally disposable processes are not too far distant and will have an increasing impact in the utilization of facilities. The advantages of such disposable processes have been mentioned; the main benefits are flexibility, reduction of set-up times and cleaning costs, as well as the possibility of high capital expense reductions. Biologics development is rapidly evolving, as is purification and filled final dosage form technology. The industry has to take advantage of such innovative technologies, as changing market and competitive conditions reduce the luxury of yield losses or idle capacities. The need for innovation in production processes is just as important as innovation in biologics drug development … and maybe even more so.
REFERENCES