The risk of contamination (especially microbiological) is always an area for special attention in biopharmaceutical processes. No matter the process stage, whether upstream of a bioreactor or in the final filling of a sterile product, effective contamination control continues to be a critical requirement, so any opportunities for improvement may justify further investigation. Even with established validated processes, demands for higher purity and increased sterility assurance may require manufacturers to reassess their procedures and technologies. New processes present an even greater opportunity to introduce innovative, enhanced technologies that can improve contamination control to satisfy regulatory requirements not only today but in the foreseeable future.
Introducing a new technology into a process may, however, bring concern over possibly time-consuming, demanding process qualification and validation studies and ultimate regulatory approval. We address these issues by using as an example new technologies that can reduce contamination risk when making aseptic connections. Here, validation data on a single-use aseptic connector is presented based on an extreme interpretation of worst-case conditions, to confirm the effective control of sterility during connection. Core validation data of this type can help make the introduction of enhanced technologies easier, faster, and safer.
Disposable Connectors and FittingsAlmost every biopharmaceutical process involves making sterile connections between fluid pathways. Such connections may be part of relatively simple systems, such as a sampling point for quality control assays or a link from a sterile fluid container to a filling lines. Traditionally, reusable fittings have been used, particularly those made of stainless steel, but the recent trend has been toward single-use disposable connectors. In some cases, connectors may be integral to more complex, single-use systems, often involving flexible bags, pre- and sterilizing or virus-removal capsule filters, chromatography purification capsules, tangential-flow filtration modules, bioreactors, and so on.
According to the Bioplan Associates’ 2006 survey of biopharmaceutical manufacturing, two primary reasons stated by biopharmaceutical manufacturers for using disposable systems were sterility assurance and reduced cross-contamination (1). Before embarking on any qualification and validation program for disposable connection devices, it is important to recognize the major features of such devices and how they may or may not be suitable for a specific process.
Major Features of Disposable Connectors: It is not our purpose here to review in detail the range of single-use connection products currently available. It is, however, important to recognize their features that can provide benefits in contamination control and would influence the content of a validation program. These connectors typically incorporate the practical benefits of quick-connect fittings, such as speed and ease of use. However, a unique feature (and one of the most important) to some recent developments is elimination of the need for connection under Grade A (Class 100, ISO 5) laminar-flow conditions. The validation approach described here is specific to these types of aseptic or sterile connectors and focuses exclusively on controlling sterility of the fluid pathway.
Aseptic/Sterile Connectors and ConnectionsMuch discussion has occurred about the semantic distinction between the terms aseptic and sterile in regards to connectors and connections. Aseptic is defined as “(a), free of pathogenic microorganisms, as in aseptic surgical instruments,” and “(b) using methods to protect against infection by pathogenic microorganisms: as in aseptic surgical techniques.” Sterile is defined as “free from live bacteria or other microorganisms, as in a sterile operating area; sterile instruments” (2).
Although the dictionary definition of aseptic leaves open the possible presence of nonpathogenic microorganisms (hence nonsterility), in practice the term is often applied interchangeably with sterile to describe the process of connecting two sterile components in a controlled (not necessarily sterile) environment, such that sterility of their interconnected pathways is maintained. In this sense, the two terms are used synonymously. As a means of distinguishing them, we can think of aseptic as describing the process of making a connection that can be validated as being sterile. So aseptic refers to the connection process, whereas sterile refers to the validated connection result.
The ability of disposable connection devices to make sterile connections in less-controlled environments places a strong dependence on the sterile barrier within the connector and on the availability of validation data to demonstrate the security of this alternative technology. Taking these concepts and features into account, we must consider how this new technology can be validated to demonstrate that it will consistently maintain a sterile fluid pathway.
Validation ApproachAt first sight, the validation approach may seem daunting to potential users of disposable connectors, especially in companies currently using traditional, manual aseptic connection methods for validated, sterile processes — as well as people who are unfamiliar with new technologies. The reality is that biopharmaceutical companies, suppliers, and organizations have addressed this task in recent years with considerable vigor and speed, resulting in publication of industry guides, recommended test methods, and generically applicable validation data. The basic framework of a validation program can now draw upon several sources.
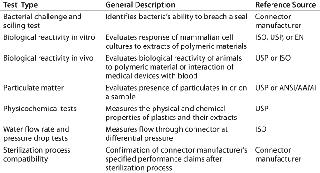
Bio-Process Systems Alliance (BPSA) Guides: Established in 2006 by the Society of the Plastics Industry to meet the challenges of emerging single-use bioprocesses, the BPSA has issued a guide on qualification test methods for connectors (3). This guide provides a good starting point for a validation program, because it describes not only the types of tests that may be appropriate but also the sources of reference tests (e.g, ISO, ASTM, USP). Table 1 summarizes the qualification tests for connectors referenced in the BPSA guide.
Table 1: Summary of qualification test methods for single-use connectors per BPSA component quality test guide
Core Validation Data from Connector Manufacturers: A substantial amount of data is now available from connector manufacturers to demonstrate the efficacy and safety of their products. Summarized in supplier validation guides, these methods and results can form a significant part of the validation support users submit to regulatory authorities.
Process-Specific Validation Data: To help confirm the suitability of a connector in a given process, vendor-supplied data may be supplemented by additional validation tests, often performed in conjunction with the connector manufacturer. In many processes, however, only a limited number of tests may be required: e.g., when simple aqueous fluids are involved or connectors form part of a presterilized disposable system with its own validation support. In such cases, a supplier’s process qualification data can form the major part of the validation package.
A speedy and successful validation is achievable by
-
following recommendations issued by connector manufacturers and organizations such as the BPSA
-
using appropriate reference tests
-
working closely with connector manufacturers and their core validation data.
The following examples provide insight into two of the most demanding tests of sterility assurance for a connector. The product tested was a Pall Kleenpak connector, a two-component connector that incorporates protective barrier peel-away strips. It is sterilizable by gamma irradiation or autoclave. After sterilization, the barrier strips protect the sterile fluid pathway before and during connection. And an irreversible locking mechanism prevents reopening.
Two functional bacterial challenge tests were performed on these connectors: a liquid challenge soiling test using Geobacillus stearothermophilus and an aerosol challenge test using Serratia marcescens. These tests were based on qualification tests described in the BPSA component quality test guide, as shown in Table 1 (3).
Functional Liquid Bacterial Challenge Soiling Test: Representing perhaps the most extreme interpretation of worst-case conditions, this test involves immersing the presterilized components of a connector into a liquid bacterial spore suspension to coat all externally exposed surfaces of each connector with >106 G. stearothermophilus spores. Because aseptic/sterile type connectors are designed to operate under dry conditions, these severely contaminated connectors are then allowed to dry before subsequent connection and sterility testing of the connected fluid pathway. Use of desiccation-resistant bacterial spores ensures high bioburden viability during connections made after drying. Full details of the procedure can be found in a published validation guide (4). Figure 1 shows simplified schematics of the test systems.
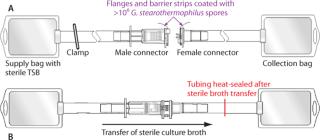
After the connection was complete, sterility of the fluid pathway was then assessed by flowing sterile bacterial culture broth (e.g., trypticase soy broth, TSB) through the connector, incubating that broth for seven days and checking for turbidity, then filtering it through a sterile 0.2-µm analytical membrane filter disc. The disc was plated onto agar, incubated for an additional seven days, and finally examined for sterility or recovered bacterial colonies.
A total of 29 tests were performed, and in all cases sterility of the fluid pathway was confirmed. Negative controls with noninoculated, presterilized connectors also gave sterile results in all tests. Positive controls (in which the protective barrier strips were removed before inoculation and connection) showed broth turbidity and confluent bacterial colonies on the analysis membrane filter discs in all tests and confirmed validity of the bacterial challenge and recovery procedure.
Functional Bacterial Aerosol Challenge Test: A second test complemented the liquid bacterial challenge soiling test in providing an aerosol challenge. We chose Serratia marcescens for this test (rather than the smaller Brevundimonas diminuta commonly used in filter challenges) to maximize sedimentation onto the connectors during use and provide a worst-case bacterial challenge environment.

This study simulated a sterile media fill similar to those performed in process-specific validation studies conducted by users. In this core validation study, however, worst-case environmental conditions for microbial levels were introduced to assess the safety margin and sterility assurance attainable when the product is used in a highly contaminated uncontrolled environment: outside a Grade A/Class 100/IS0 5 environment or even a Grade C/Class 10,000/IS0 7 or Grade D/Class 100,000/ISO 8 environment. Figure 2 provides simplified schematics of the test systems.
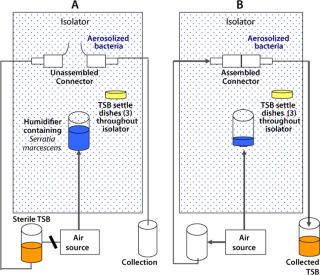
After exposure of unassembled sterilized connectors (binding controls) to aerosolized bacteria, the connectors were joined, and sterile TSB was passed through them into a collection vessel. The liquid was then incubated and assayed for bacterial content, as described above. Suitable positive controls were included, with their peel-away strips removed before the aerosol challenge, as well as negative controls with no aerosol challenge. Table 2 summarizes the results obtained.
Table 2: Serratia marcescens aerosol challenge test on Kleenpak connectors

Aerosolized bacteria conditions represent a severely uncontrolled environment exceeding Grade D/Class 100,000/ISO 8 particulate levels by >100× and microbial aerosol levels by >10,000×. In all tests, sterility of TSB transferred through the completed connections was maintained. The negative controls (sterile media transferred and filled with no aerosol bacterial challenge) also gave sterile results in all tests. And as expected, the positive controls (with protective barrier peel-away strips removed before bacterial aerosol challenge and connection) gave bacterial counts (nonsterility) in the transferred TSB.
The practical significance of these aerosol challenge data for pharmaceutical manufacturing environments can be demonstrated by comparing these results with environmental microbial limits in GMP guidelines. The levels presented in Table 2 are more than 106× higher than the maximum permitted microbial limits for Grade A/Class 100/ISO 5 environments, according to EU Annexe 1 and US aseptic processing guidelines (5, 6). As noted above, the levels are also more than 104× higher than Grade D/Class 100,000/ISO 8, the environment in which these connectors are designed to function. So we concluded from the bacterial aerosol challenge functional test that sterile fluid pathways can be maintained during connections, even under worst-case environmental conditions.
Easy, Fast, and SafeReducing the risk of bacterial contamination and achieving high sterility assurance levels are major driving forces behind the application of disposable connectors. In 2002, Richard Friedman of the US FDA’s Center for Drug Evaluation and Research (CDER) presented data showing a substantial increase in product recalls associated with lack of sterility assurance over a three-year period (7, 8). At the same time, the FDA also recognized that “enhanced technologies” such as isolators and form–fill–seal machines could contribute significantly toward improving sterility assurance. Indeed, revisions or appendices were specifically added to subsequent FDA and EU GMP guidelines in recognition of the special nature of these technologies and to encourage their use wherever applicable.
Manufacturers of enhanced aseptic processing technologies, which include single-use aseptic/sterile connectors, can assist by providing comprehensive validation data on the functionality and safety of their products. The data presented here constitute an example of core validation studies which, together with other published data, can help to make the introduction of such new technologies into processes easier, faster, and safer.