 Advanced-therapy medicinal product (ATMP) characterization and analysis play important roles in providing chemistry, manufacturing, and controls (CMC) information for regulatory applications as well as in supporting product-release and stability studies. Each type of advanced therapy presents different analytical development challenges, so each requires specific characterization, potency, purity and identity assays. Variability in cells and among patients, multiple and complex mechanisms of action (MoAs), a general lack of readily available reference materials, and complicated analytical methods and instruments underlie the major technical difficulties (1).
Advanced-therapy medicinal product (ATMP) characterization and analysis play important roles in providing chemistry, manufacturing, and controls (CMC) information for regulatory applications as well as in supporting product-release and stability studies. Each type of advanced therapy presents different analytical development challenges, so each requires specific characterization, potency, purity and identity assays. Variability in cells and among patients, multiple and complex mechanisms of action (MoAs), a general lack of readily available reference materials, and complicated analytical methods and instruments underlie the major technical difficulties (1).
Many techniques for analyzing cell-therapy products are not yet recognized as standardized pharmacopeial methods. Regulatory requirements are evolving around the world, and further guidance documents are expected for both the United States and Europe in the near future. Thus, product validity should be demonstrated for regulatory authorities through assay validation, the extent of which depends on the product-development stage.
Below, I answer the five “W” questions — who, what, when, where, and why — to discuss the testing strategies used by cell-therapy manufacturers throughout their products’ development lifecycles. Addressing the “how” then highlights some commercial challenges often encountered, such as method validation and potency assay development.
The Five Ws
Who and Where: At Pluristem, we have a quality control (QC) laboratory that complies with current good manufacturing practice (CGMP) requirements. The laboratory supports development of different products from early stages through to in-process control and product release assays for clinical trials. The QC laboratory is staffed by qualified scientists who are experts at developing, optimizing, qualifying, and validating analytical methods as well as transferring those methods from research and development (R&D) into a GMP QC laboratory. Other qualified scientists are skilled in routine testing, and they participate in method-validation efforts.
What: Our labs test for product quality, identification, potency, impurities, and safety. We have in-house assays for recovery, dose viability, cell size, and population doubling, the Vi-Cell counting instrument from Beckman Coulter being useful here. In addition, we test for a number of metabolites during cell growth — glucose, lactate, ammonia, glutamine and glutamate, and lactate dehydrogenase (LDH) — using a Cedex-Bio instrument from Roche.
We also have identification assays for our mesenchymal cells based on immunophenotyping with a CytoFlex flow-cytometer system from Beckman Coulter. This instrument helps us to determine cellular characteristics and detect specific markers.
We measure the composition of our batches using a sophisticated method based on DNA fragment analysis of microsatellite sequences that are polymorphic in length from person to person. For this we use quantitative fluorescent polymerase chain reaction (QF-PCR). Elsewhere, the same method is used in a range of applications, including genetic mapping for autosomal recessive disorders, detection of maternal-cell contamination of amniocentesis samples, forensic DNA fingerprinting, and paternity studies.
Pluristem relies on in-house, cell-based potency assays using different cell lines on the Synergy HT fluorimeter from BioTek Instruments. Potency assays are critical in assuring the quality and consistency of the product. We also run enzyme-linked immunosorbent assays (ELISAs) on a Synergy HT ELISA reader from BioTek Instruments to measure secretion of cytokines, interleukins, and monocyte chemoattractant proteins.
In addition we have in-house impurity assays. We test for process-related impurities (e.g., residues of fetal bovine serum and enzymes) as well as product-related impurities (e.g., blood cells or hematopoietic cells from the placental source) based on immunophenotype.
Our safety testing is performed mostly by external laboratories that monitor for sterility, mycoplasma, donor eligibility, and virus contamination. Those laboratories must have the highest accreditation standards. When products are made for the US market, for example, we use a laboratory approved by the US Food and Drug Administration (FDA).
Why: Analytical method development and validation are critical to obtaining the reliable analytical data that we need to reach our development milestones. And coupling a good understanding of current regulatory expectations and relevant methods with advanced instrumentation is critical to developing efficient, accurate, and reliable analytical methods.
Regulators expect companies to use cell counting as an in-process control (IPC) for yield and also for calculating passage number against population-doubling levels (PDLs). To ensure that we’ll have sufficient amounts of cells for further production, we use this method to verify the population of each vial in our cell bank. The PDL acceptance range should demonstrate that no such difference between batches will compromise functionality or characteristics of cells at the end of production.
Genetic stability testing (karyotyping) also is expected for cell banks/stocks. Regulators want to see precise methods — e.g., comparative genomic hybridization (CGH) or fluorescence in situ hybridization (FISH) — used to supplement standard karyotyping, at least as part of this characterization effort.
All analytical methods used for IPC and for release and stability testing must be validated. That should be documented clearly, and validation of analytical methods should be conducted according to ICH Q2 (2).
Antibiotics normally are not recommended during cell therapy processing because their effects can “mask” contamination and/or compromise sterility-test performance. Traces of antibiotics in a final product would be classified as clinically relevant impurities, and you may be requested to test every product batch for those.
Potency assays should be part of stability testing because their stability-indicating results reflect the biological activity of an active substance. Each batch should be measured for potency against that of a reference batch, and the related specification should be based on historical data. Potency assays should identify subpotent batches, and the results should correlate with clinical efficacy.
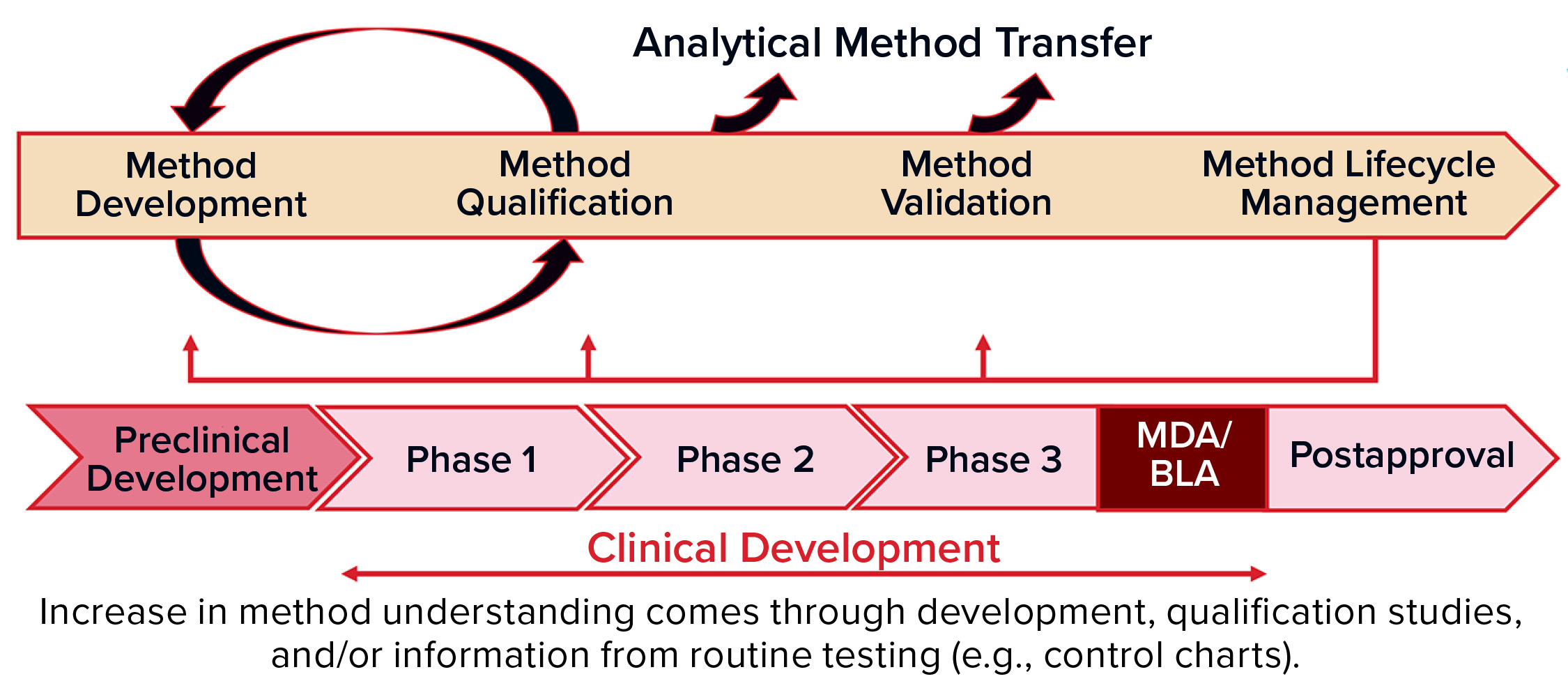
Figure 1: Methods validation requirements and analytical methods evolve over the commercial lifecycle, from early stage development through to commercial use.
When: Analytical method development and validation should evolve through the commercial life cycle of a product (Figure 1), from early stage development through commercial use. You should begin to develop a method during preclinical stages and optimize it through phase 1 clinical testing up to phase 2. Then you can qualify the method and validate it for phase 3. During all those phases — and after marketing, of course — you will be performing routine testing with control charts to monitor for drift in the manufacturing process and/or analytical methods.
When you are in the preclinical stage or phase 1, you focus on developing and validating safety assays. But for potency assays and others, you’re just beginning with feasibility. And then over time, you will continue with optimization of those methods and develop understanding about your critical reagents. You’ll establish standard operating procedures (SOPs) and system suitability documentation, and you’ll choose your reference standards. During routine QC testing, you can determine whether improvements are needed. If so, that leads to assay optimization.
In phase 2, you’ll have some data from QC GMP testing to help you challenge your ranges and move into robustness and a qualification. And in phase 3, you should know much more about your assay and be ready for validation. All that time (especially phase 2–3 and post marketing), monitoring analytical trends will show how your reference standard is working and how your production batches turn out to monitor for drift in the method, reference standards, or process. You should be hands-on throughout. If something changes, you have to revalidate depending on the extent of those changes and cost of doing so.
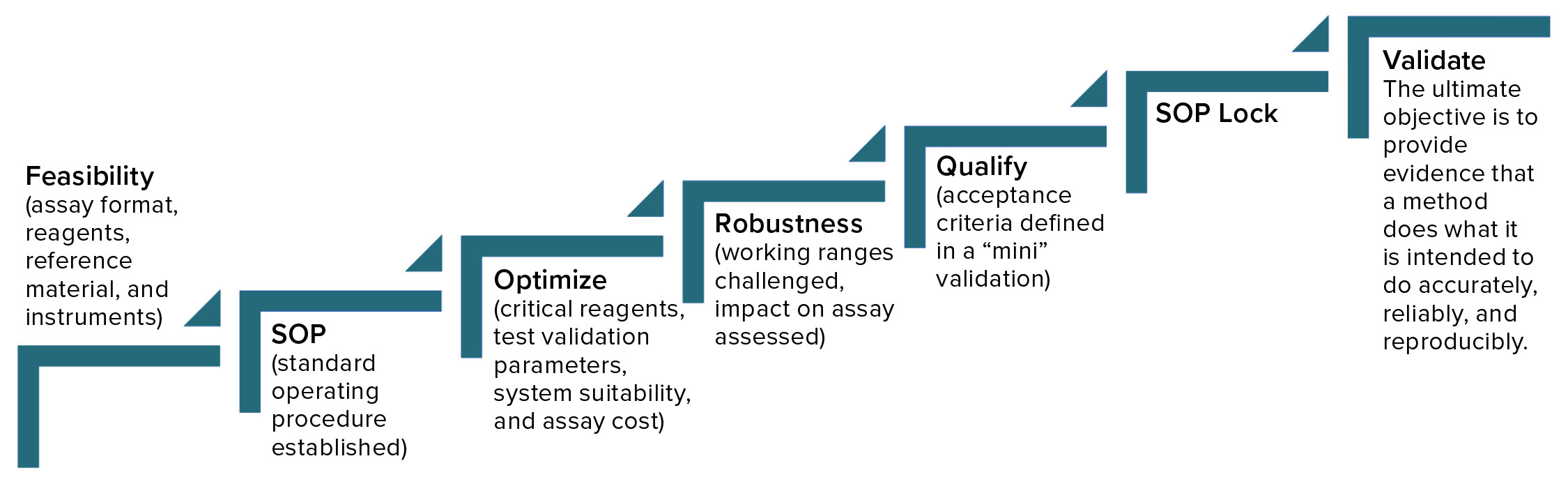
Figure 2: Analytical method lifecycle management
How Do We Develop an Assay?
Begin with feasibility (Figure 2). Start with understanding (the type of method, its intended use, and relevant regulations), and then develop a method, keeping its validation parameters in mind. You’ll begin with an assay format, choosing reagents and references, identify the necessary instruments, and then establish a SOP, which you will optimize continuously going forward. It’s important to determine which will be the critical reagents, what parameters of validation you will need to address, and establish system suitability. You also have to consider the cost of a given assay, which you can reduce by changing reagents or detection methods, for example. It’s good to address robustness early during development and then during qualification, and validation, when you can challenge working ranges and assess their impact on assay results.
Method qualification is like a “mini” validation study and is very important to helping you determine when an assay is ready for validation. It helps define the acceptance criteria, beginning with broad ranges that will be narrowed down during validation. At that point, you’ll lock in the SOP, so the process itself needs to be locked in already. The ultimate objective of validation is to provide evidence that a method does what it is intended to do accurately, reliably, and reproducibly. Depending on the type of method involved, you will have to test for a number of validation parameters. For example, if a method is used only for product identification, then testing that method for specificity alone is enough. But for potency assays, you should test more parameters (Table 1): accuracy, range, linearity, precision, and possibly quantitation limit.
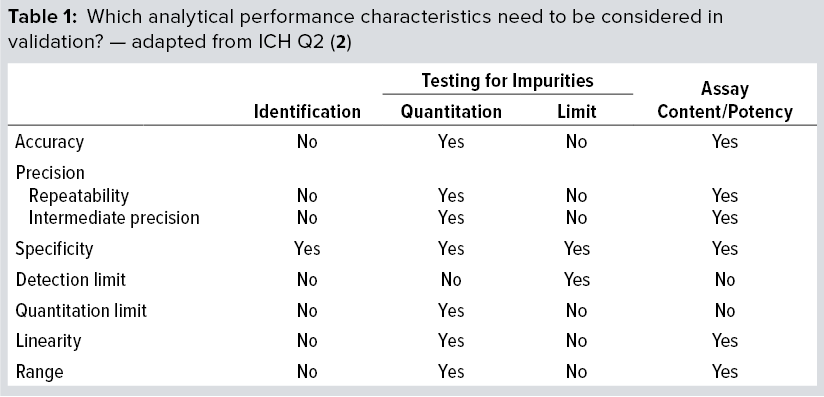
Challenges Faced in Developing a Cell-Based Potency Assay: Measurement of potency plays an essential role in quality control, GMP product release, comparability, and stability testing for both drug substances and drug products. Biological activity is a critical quality attribute (CQA) that often faces more regulatory scrutiny than other parameters receive. Potency should be specific to a biologic’s MoA and validated according to ICH Q2(R1) standards (2).
We use several cell-based assays at Pluristem for testing cell proliferation and inhibition, cell migration, and cell secretion. Such assays can determine the relative potency of a product by comparing its biological response (activity) — related to its MoA — with that of a control/reference preparation usually based on pharmacopeial or in-house standards.
Several factors can contribute to cell-based potency assay variability. Therefore, the sources of assay variability should be considered and limited as much as possible. Instrumentation is a common example. Assay variability is a particular concern when you use sophisticated instruments. They need to be qualified, and the people running the assays should be qualified as well.
Pluristem’s PLX-R18 product could address an unmet need in patients transplanted with hematopoietic cells who do not respond to stimulating factors or hormones to induce production and release of the blood cells in which they have been deficient. PLX-R18 is designed to stimulate the regenerative potential of bone marrow to produce blood cells. Thus, one potency assay for the product measures bone-marrow migration. The test uses a transfer plate on which bone-marrow cells are seeded in the upper end and conditioned medium is added to the lower end, so the cells should migrate from one to the other. Cell-counting technologies can differ in their accuracy, which could affect activity calculations. So for this assay, we had to decide whether to use a direct method (enumerating bone-marrow cells in migration with a cell counter) or an indirect method (using a fluorescence-based plate reader to quantify bone marrow that has migrated).
For the direct method, cell counters should be qualified — installation, operation, and performance qualification (IQ/OQ/PQ) — and well maintained. Cell counting is a relatively inexpensive and straightforward detection method. But it doesn’t measure low numbers of cells (in migration) as accurately as large numbers (e.g., total cells from a tested sample). So we would need to use a very large number of bone-marrow cells in a large-scale assay for the best results.
That makes an indirect method worthy of consideration, and the detection method becomes the challenge. We can use an endpoint assay that measures DNA content in cells or tests for their metabolic activity (e.g., the alamarBlue test). Filter sets, instrument gain, and microplate types all are important variables to keep in mind with fluorescence-based detection. We need to confirm the excitation and emission maxima based on a given fluorophore with an appropriate filter and set the instrument gain appropriately. Clear-bottomed plates enable us to analyze cell morphology before running the assay, but they can increase crosstalk. Alternatively, black-sided well plates can reduce scatter, signal overlap, and crosstalk.
Reagents are another factor that can contribute to variability of cell-based potency assays. All reagents in a GMP laboratory should be qualified, and you should know which present higher risk for a given use. Lot-to-lot variability of critical reagents can cause variability in test results. So each reagent must be assessed for its criticality level, with the results documented. You will have to screen every new lot of the critical reagents, whereas simple verification is enough for low-risk ones.
For cell-based assays, the cells themselves are like critical reagents. Choosing the right cell type to study is a key factor in successful method development. Different types (e.g., primary cells or cell lines) generate different biomarkers, which can influence assay results. Note that target cells should be grown and stored in the same way as origin cells (during feasibility studies). Cell-line stability and cell identity are both important to preserving assay repeatability over time. Primary cells such as hepatocytes, bone-marrow cells, and endothelial cells are good options for in vitro studies, but they are all difficult to grow and can contribute to assay variability.
We use primary bone-marrow cells in our potency assay. They’re collected fresh from mice, and because we would have to use a large number of them with a cell-counting instrument, an alternative is to choose an indirect measurement instead and 96-well Transwell plates to reduce the volume of cells and reagents needed. Bone-marrow cells do not expand in culture, so a great number must be banked for future use. If you cannot have a stock bank, then assay repeatability will be compromised. So it’s important to qualify your working bone-marrow cells and categorize them as critical reagents. Each new cell stock will be screened against the current stock. We also tested different incubation times to determine the best ones for migration measurement.
Because sparsely cultured cells can have poor biological activity, and high-density cultures can cause inaccurate results (when cells are in a plateau phase), consistent and optimized cell density is important for accurate determination of assay range. A matrix of different cell numbers is generated to find the most accurate and precise range. High-density cultures also can cause cells to detach from the plate wells; attachment factors can be added to help mitigate that effect.
Plate-edge effects also can contribute to variability of cell-based potency assays. Cells in wells around the edge of a plate can react differently to assay conditions than those in the inner plate wells because of temperature differentials across plates in an incubator. We don’t want to stack them up because perforations can form during long incubation periods. I recommend running duplicate or triplicate samples (to randomize those plates) and monitoring time, plate order, and plate position in the incubator. Do your best to ensure that incubator temperature is distributed evenly.
We calculate relative potency based on a reference standard, which is both an established scientific principle and a regulatory requirement. Our reference standard is a representative batch produced by the relevant manufacturing process, so each product will have its own reference standard. It is essential that tests of standards and samples be carried out at the same time and under identical conditions on the same plate.
Regulators expect reference batches to come from the best-characterized lots available that represent your current manufacturing process. Reviewers will want to see a protocol in place that shows how new reference materials are qualified for use and how you prevent drift over time when each new reference material is qualified against the previous one. Extended characterization is expected in addition to standard release testing for each reference material. And stability of reference materials should be verified through stability testing.
| Cell-Based Assay Validation |
| Study Design: Four analysts ran six different experiments on different days using different lots of critical reagents.
Repeatability: ≤13% relative difference (RPD) Intermediate Precision: ≤7% coefficient of variation (CV) Accuracy: ≤14% relative error (RE) Linearity: R2 ≥ 0.95 Range: 70–130% practical range (Rp) |
We use reference standards for system suitability testing; defining assay acceptance criteria (positive control); assay development, qualification, and validation; determining relative biological activity; testing/screening of critical reagents; and monitoring method drift over time. For bridging and screening critical reagents, I choose several lots to use in a given assay. A company should have an SOP that describes how to identify critical reagents based on their complexity. For example, biologic reagents are more complex and pose more risk than chemical synthetic reagents. Screening them means running the reagents in the assay for which they’re intended — with documented acceptance criteria to evaluate them.
Fetal bovine serum, for example, is a critical reagent both in the laboratory and in cell-therapy manufacturing. We test it using several assays for which it is a critical reagent, and we’ll compare the results against those from a laboratory standard. We’ll buy several lots and use the one that gives us the best comparison to our current reagent.
In choosing reagents, we must ensure not only that they perform well, but also that their manufacturers can assure supply at least for a year. We have to consider the expiration dates and compare performance of fresh reagents with those that have been stored.
Figure 3a shows an example control chart from a product reference standard in one of our potency assays, with the normal dose distribution, and the “Validation” box summarizes the results for our cell-based assay. This example shows that if you develop and optimize a method and then qualify it well, then you can get very good results for a biological assay. Relative-potency results (Figure 3b) illustrate the analytical trend of batches that we tested at Pluristem using this assay. It mostly remains in the normal ranges, but any red dots that appear out of those ranges would be batches that get flagged as out of specification (OoS).
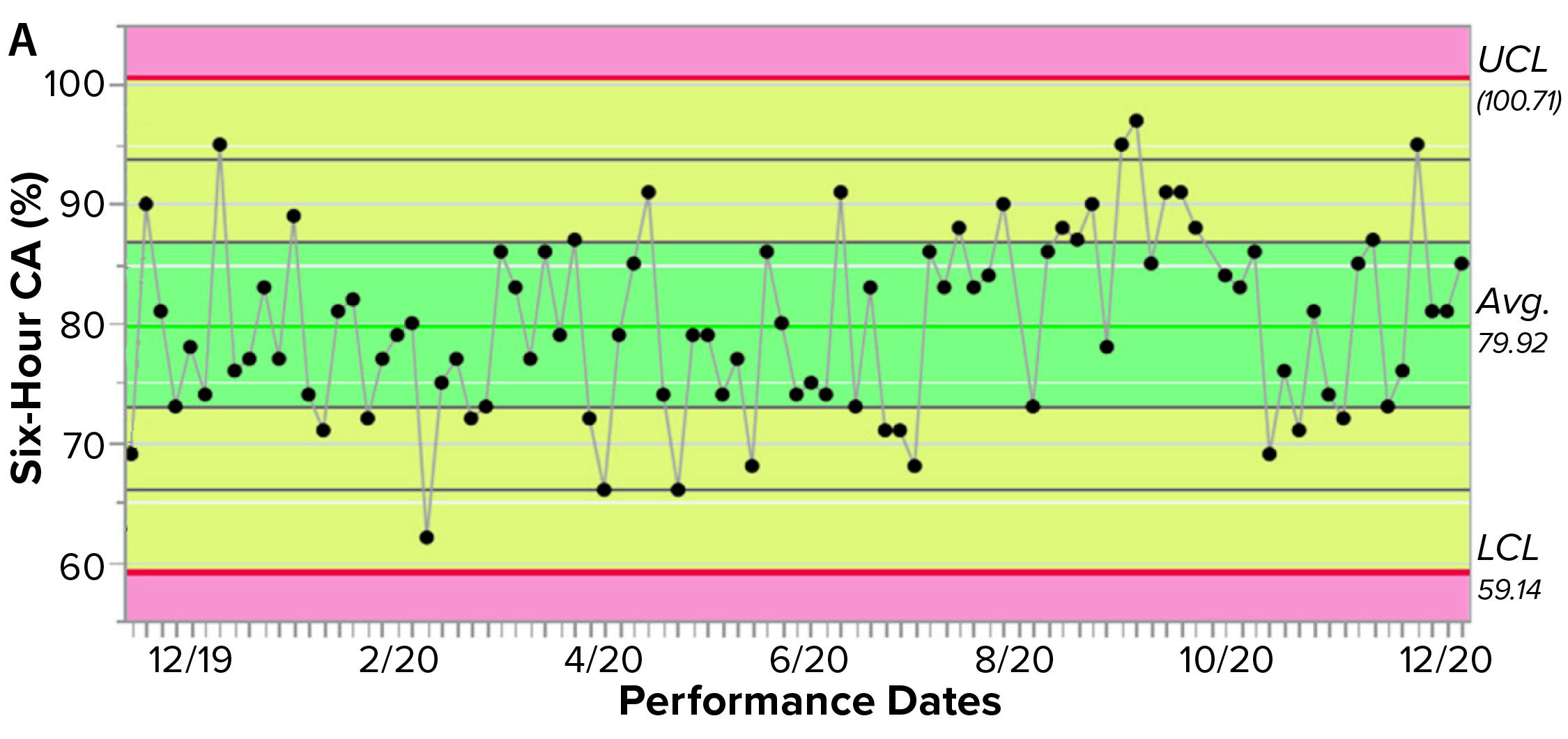
Figure 3a: Control chart and relative potency results for expanded placental cells; (a) shows results from different cell-adhesion (CA) assays (UCL = upper control limit, LCL = lower control limit).
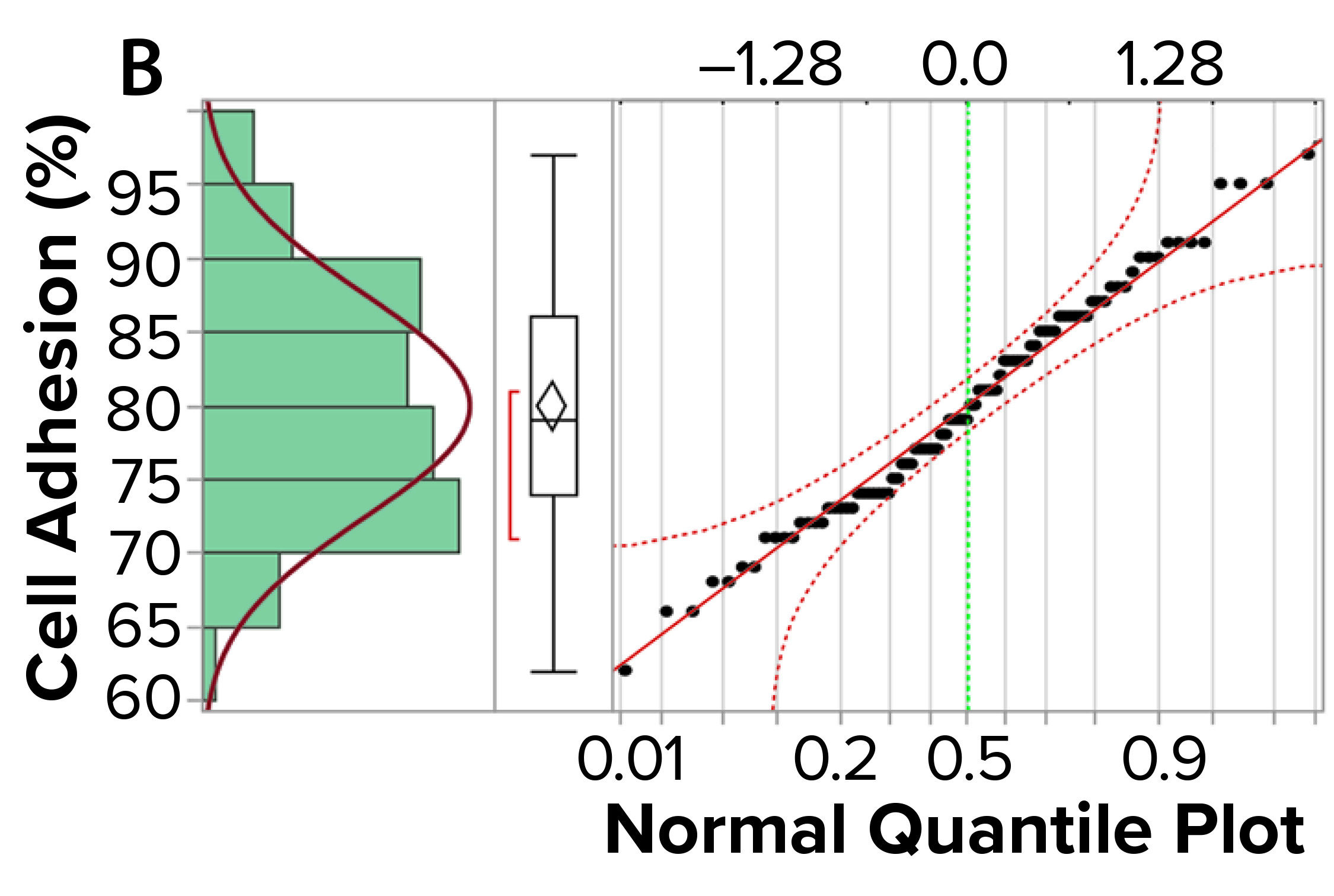
Figure 3b: Control chart and relative potency results for expanded placental cells; (b) plots cell-adhesion results distribution
Assay Robustness: A common weakness that comes up in validation of cell-based potency methods is that they are not robust enough. If you can build robustness into methods early in development, then they are likely to be efficient for quality testing. Robustness describes the capacity of an analytical procedure to go unaffected by small but deliberate variations in method parameters, thus providing an indication of the test’s reliability during normal use.
Experimental design of robustness studies can be conducted according to a scientist’s univariate approach (changing a single variable or factor at a time) or a statistician’s multivariate-analysis approach (studying the effects of multiple variables on a process simultaneously). Using the former approach with our bone-marrow assay, we tested the range of incubation times for product cells, then the range of migration times for bone-marrow cells, and finally the incubation times of the latter with reagent detection. Statistical analysis of the results provided a means for narrowing down assay variability.
Immunophenotyping: To characterize the membrane markers on our mesenchymal cells, we use a CytoFlex instrument. It has three lasers, nine fluorescent channels, and very high sensitivity, with a user-adjustable flow rate (10–240 µL/min) and a plate loader for standard V-bottom plates. That makes it a sophisticated new technology that provides high sensitivity and unique compensation workflow while increasing efficiency.
The immunophenotype method accurately and precisely elucidates the percentage of mesenchymal cells that present typical cell-surface mesenchymal biomarkers: CD105, CD73, CD29, and CD90. We also need to detect “negative markers” that indicate impurities (e.g., hematopoietic or other cells). Using the CytoFlex instrument, we can quantify impurities accurately to a very small lower limit of quantitation (LLoQ).
Instrument qualification is the foundation of good data. The environment, utilities, electrical components, and hardware all should be verified and documented as conforming to vendor specifications. For OQ and PQ of the CytoFlex instrument, we focus on optical precision, system alerts, software functionality, detection efficiency, acquisition carryover, and system suitability for our analytical method.
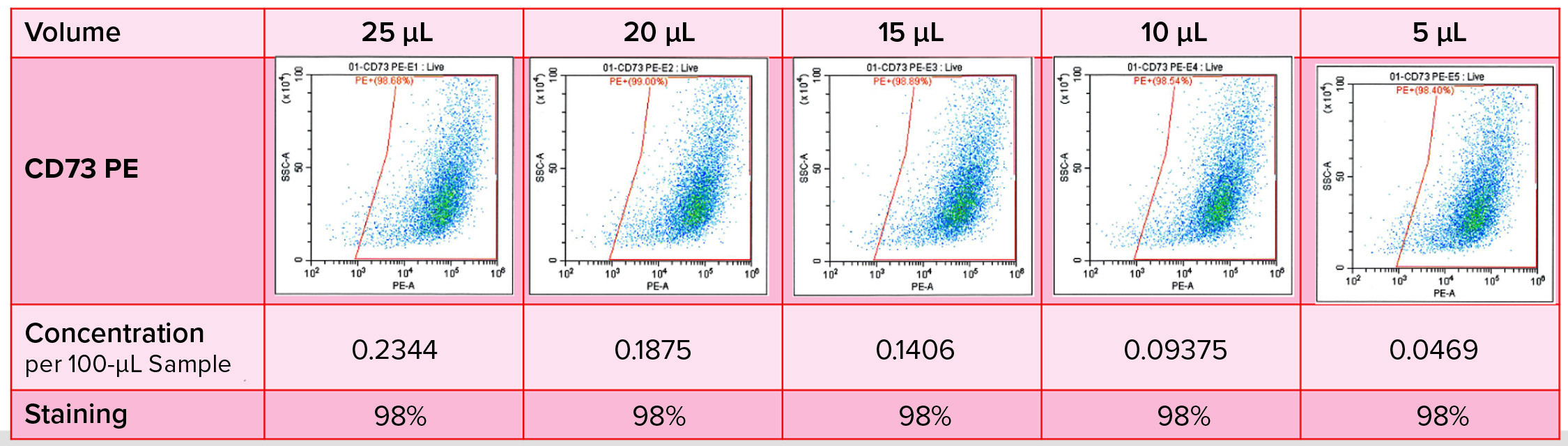
Figure 4: Immunophenotyping optimization — titration of antibodies
Antibody titration is important in optimization of this immunophenotyping method (Figure 4). We had to determine specificity and staining intensity of the antibody and minimize compensation requirements while determining the quantity of antibody to be used, enabling the method to serve as a quality control for each new antibody load. In developing such a method, we consider several parameters: e.g., sample preparation, elimination of nonspecific binding, antibody titration, detection and elimination of dead cells, dye stability, fluorochrome choice, and the acquisition template and gating strategy.
| Immunophenotyping Validation |
| Positive-Marker Study Design: Two analysts ran six different experiments on different days using different lots of critical reagents.
Range: 80–100% of cells expressing each biomarker Repeatability: <5% coefficient of variation (CV) Intermediate Precision: <5% CV Accuracy: ≤10% RE Linearity: R2 ≥ 0.95 Robustness: No deterioration was observed over four hours. |
The second “Validation” box summarizes our results for the immunophenotype method covering positive markers CD105, CD73, CD29, and CD90. We mixed stained and unstained cells to show the range of 80–100% of cells expressing that marker. And we realized good precision here, with a coefficient of variation (CV) of <5%, as well as accuracy and linearity in that range.
Next, we validated for negative markers (impurities) CD45, CD14, CD19, GlyA, CD34, HLA-DR, and CD31 (data not shown). We spiked positive controls (blood cells) into our mesenchymal cells at a low percentage and obtained good precision in the end.
The Value of Teamwork and a
Holistic Strategy
Analytical methods are essential tools for control of cell-therapy manufacturing processes, raw materials, intermediates, and final products. Typically such methods are developed by R&D groups, and then validation becomes the responsibility of quality assurance (QA) and quality control (QC) groups. It’s important that those groups work together as one team. Transfer of analytical methods from one group to another is an important step in ensuring that the proper validation will be in place to justify their intended use.
References
1 Wiwi C. Analytical Considerations for Cellular Therapy Manufacturing. Cell Gene Ther. Ins. 2(6) 2016: 651–661; https://insights.bio/immuno-oncology-insights/journal/article/448/analytical-considerations-for-cellular-therapy-manufacturing.
2 ICH Q2(R1): Validation of Analytical Procedures: Text and Methodology. US Fed. Reg. 62(96) 1997: 27463–27467; https://database.ich.org/sites/default/files/Q2%28R1%29%20Guideline.pdf.
Further Reading
Bohn J. 10 Tips for Successful Cell Based Assays. Enzo Life Sciences: Farmingdale, NY, April 2015; https://www.enzolifesciences.com/fileadmin/redacteur/bilder/landing_pages/
TechNotes/2015/Enzo_10-Tips-for-cell-based-assays.pdf.
Bravery CA, French A. Reference Materials for Cellular Therapeutics. Cytother. 16(9) 2014: 1187–1196; https://doi.org/10.1016/j.jcyt.2014.05.024.
Ruti Goldberg, PhD, is compliance and methods validation manager at Pluristem Therapeutics, MATAM Advanced Technology Park Building #5, Haifa 3508409, Israel; 972-74-710-8600; rutig@pluristem.com; https://www.pluristem.com.
This article is adapted from a presentation at Informa Connect’s Cell and Gene Therapy Manufacturing and Commercialization Digital Week in February 2021. Find more information about upcoming Informa Connect events at https://informaconnect.com/bioprocessing-manufacturing/events.