Biotechnology pipelines have demonstrated significant growth over the past decade, with many therapeutic candidates evolving in a single class of protein molecules: the monoclonal antibodies (MAbs). To develop such therapeutic candidates, a scalable drug development process must leverage in-house and industry-wide knowledge so biotechnology companies can address the economic and medical needs of 21st-century medicine.
Biotherapeutics development is complex, resource intensive, and time consuming, taking some 10 years of effort to go from target validation to commercialization. This reality, coupled with rapid technological advances and evolving regulatory expectations, limits the ability of biotechnology companies to progress rapidly with their pipeline candidates. Also, the biopharmaceutical industry is heavily regulated, but most regulations are targeted at commercial products, with a significant gap in available guidance for earlier stages of product development.
This two-part article discusses the analytical activities associated with the progression of biotherapeutic candidates from the early stages of clinical development through their appearance as licensed drugs on the market. The analytical program for a given biotherapeutic has a lifecycle analogous to that of a manufacturing process used to prepare material for clinical and commercial use. Here we examine the stages it goes through, and Part Two will provide more detail on method qualification, validation, and remediation.

The lifecycle of analytical methods for biotechnology products can be divided into two distinct phases: a developmental phase (activities that span initial method development through use in a clinical program) and a validated phase (late-stage activities for methods used in commercial settings). Activities associated with the former are extensively described in scientific publications, whereas the validation phase is described in regulatory guidance documents such as those contained in the US Code of Federal Regulations (CFR), US Pharmacopeia (USP), and International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH).
An ICH guidance document covers validation activities for product registration, so the guidance is specifically applicable to commercial products (1). Although no specific guidance on method validation is available for earlier stages of product development, industry practices have evolved to provide assurance of acceptable method performance in such earlier stages. The generally accepted term for precommercial activities is method qualification, but the scope of that qualification, its timing with respect to the stages of product development, and its relationship to validation activities has not been consistently delineated or practiced.
To organize qualification practices and requirements into a single logical construct, the spectrum of analytical activities can be thought of in a lifecycle model using the following fundamental principles:
-
The analytical lifecycle can be divided into well-defined segments that relate to each specific stage of product development. Organizing the analytical lifecycle this way provides a basis for consistency and scalability because it applies to multiple methods for multiple development candidates and commercial products.
-
Method validation is considered the pivotal point in this lifecycle because it justifies use of a method in commercial settings. In addition, this activity defines the point at which ownership of methods transfers from an analytical development group to a quality group. These activities are typically initiated after a sponsor has committed to commercializing a drug candidate (generally after positive feedback from first-in-human clinical trials).
Figure 1 details the process described in brief above. It applies to analytical methods destined for a GMP environment, in which they will be used to make product disposition decisions, including release testing and stability studies. This process is not intended for characterization methods, which are generally carried out within an analytical development organization and used to characterize a protein. The process encompasses several distinct activities, some of which are carried out both in early development (initial, subscript i) and later in development to enable validation (subscript v), as follows:
-
Method development and improvement
-
Method robustness studies (Ri and Rv)
-
Establishment of system suitability (SSi and SSv)
-
Method qualification (Qi and Qv)
-
Method validation (MV)
-
Method remediation (after product licensure).
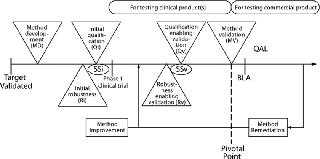
Typically, analytical method development (MD) begins after a biological target has been validated, a protein therapeutic has been defined (primary sequence), and a sponsor has decided to initiate human clinical trails. If a method is intended for use in release and/or stability testing of clinical material, its suitability for the intended purpose is demonstrated by initial qualification (Qi). Later in development, typically right before pivotal phase 3 clinical trials begin, method developers perform qualification studies for MV: validation-enabling method qualification (Qv). Qi and Qv are accompanied by corresponding robustness studies, initial robustness (Ri) and validation-enabling robustness (Rv). In addition, when method development is finished, initial system suitability (SSi) should be established. Reassessment and refinement of system suitability performed for validation (before commercialization) is annotated as SSv.
MV is typically completed before process validation in compliance with CGMP procedures outlined by ICH Q2R1 (1). Before MV, all activities are typically “owned” and led by the group developing the method, whereas validation and postvalidation activities are typically “owned” and led by quality. However, this process provides flexibility in assigning ownership as deemed necessary. Its modularity allows for independent execution of activities that can be separated in time or location. For example, discrete packets of activities can be parceled out among subgroups in an organization as resources allow without affecting the overall process integrity.
Method DevelopmentExtensive literature is available on best practices in analytical methods development, typically focusing on pitfalls related to each specific methodology, technique, or instrument. A common industry practice is to provide in-house guidance documents on these topics. Following such publications and guidelines can significantly speed up development. Some larger biotechnology companies recently have been implementing “platform technologies” to effectively deal with their growing pipeline of products in the same class (e.g., MAbs). A platform approach provides for use of the same (or similar) analytical methods for evaluating quality attributes of many products, facilitating rapid development of new drugs while allowing for evaluation of process- and product-related impurities.
During biopharmaceutical development, a wide variety of analytical technologies are used to assess a product’s physicochemical and functional characteristics. As development proceeds, many such tools evolve into analytical methods for routine evaluation of product-quality attributes. Proteins are not structurally homogeneous, so a single peak in a chromatogram, electropherogram, or spectrum may not represent a single molecular entity (as is typical for small molecules). No single analytical method unequivocally defines product purity; therefore, a combination of methods is typically used to properly describe product purity.
MD should conclude with a documented analytical protocol (typically a method development report) that specifies a method’s final operating conditions. It is also important to document development history with scientific guiding principles underpinning the method and data gathered. Method development typically concludes with robustness and qualification studies, which should also be documented either as part of the MD report or in a separate method qualification document.
Method Improvement: If justified, MD may be revisited during development of a commercial manufacturing process. In such cases, redeveloped (improved) methods should follow the typical process for demonstrating suitability. Improvements are usually triggered by needs to improve resolution and/or reliably quantify isoforms/impurities that become evident as product understanding increases. In such cases an assessment determines whether a brand new method needs to be developed or whether the scope of the existing method can be expanded to cover new requirements. Either way, appropriate qualification activities, robustness studies, and (if necessary) establishment of modified system suitability criteria must be carried out.
Stability-Indicating Properties: Methods used to assess drug substance (DS) and drug product (DP) stability should be demonstrated to detect changes in quality attributes. This can require forced degradation studies on appropriate sample types using conditions known to affect protein quality (e.g., elevated temperature, pH extremes, and incubation with oxidizing agents such as hydrogen peroxide) to induce molecular changes such as aggregation, deamidation, peptide bond cleavage, and protein oxidation.
Sample stability also plays an important role. Testing for sample stability encompasses two separate assessments: evaluation of sample storage conditions before analysis and stability assessment of prepared samples awaiting analysis.
Stability and handling of samples should be established for each method by its developer before GMP testing. Samples are often stored frozen after collection and thawed for analysis. In such instances, sample integrity should be assessed over a minimum of one freeze–thaw cycle for each sample type (preferably more than one cycle). Sample stability after preparation and before analysis (e.g., time spent in an autoinjector) should be evaluated to determine an assay’s maximum duration. Details of sample handling should be included in each testing protocol.
System SuitabilitySS testing is an integral part of every analytical method and should be designed during method development. This testing demonstrates that all constituents of an analytical system (including hardware, software, consumables, controls, and samples) function as required to assure test result integrity. SS should be demonstrated throughout an assay by analysis of appropriate controls at appropriate intervals.
Existing guidance can be found in ICH Q2 and USP <621 > ; they were developed for pharmaceutical compounds and may not be directly applicable for structurally complex and inherently heterogeneous proteins, which require additional considerations beyond those typically required for small molecules. For example, appraising resolution for an ion-exchange column by measuring the number of theoretical plates (commonly done for small molecules) may not be the best way to assess system readiness to resolve charge isoforms. However, that methodology may be a good indicator to measure system performance for size-exclusion chromatography (SEC).
To appropriately establish SS, consider both the parameter that will be assessed and its associated numerical or logical value(s), generally articulated as acceptance criteria. SS parameters are the critical operating parameters to identify an analytical method’s performance. They can be established prospectively for each class of methods. It is a good practice to establish them before qualification and to demonstrate that they adequately describe the operational readiness of a system with regard to such factors as resolution, reproducibility, calibration, and overall assay performance.
SS should be tracked from the earliest possible time in the development. Tracking SS provides important information about the evolution of a method as well as the value of a particular parameter for assessing initial system suitability (SSi). Initial qualification experiments (Qi) should be used to determine average values and standard deviations of those parameters before a method is routinely used in product disposition. Before validation, SS parameters and acceptance criteria should be reviewed to verify that previously selected parameters are still meaningful — and to establish their limits such that meaningful system suitability for validation (SSv) is firmly established.
Method RobustnessMethod robustness demonstrates that the output of an analytical procedure is unaffected by small but deliberate variations in its parameters. Initial robustness (Ri) and robustness-enabling validation (Rv) are key elements of analytical method progression connected to corresponding qualification studies (Qi and Qv, respectively). These studies can be deemed successful if phasespecific robustness studies have been successfully executed. It is good practice to prospectively establish a general design (outline) for such studies. Typically a reference standard and/or other appropriate samples are analyzed at the nominal load.
Method robustness experiments cannot start before the final conditions of a method have been established. It is a good practice to identify operational parameters and divide them in order of importance into subcategories. We divide the operational parameters of different methodologies into four categories:
“Essential” category parameters are critical to method output and therefore require evaluation.
“Less important” category parameters are not as critical as those in the above category, but that may still affect the output; should be evaluated at a scientist’s discretion.
“Depends on Method” category parameters may affect method output differently for different methods and thus should be treated differently for each.
“Not useful” category parameters are known to have no affect on the output and need not be evaluated for robustness.
Table 1 shows our current thinking on the importance of operational parameters for separation techniques. Categorizations can be revised as existing technologies advance and new technologies become available.
Table 1: Categorization of operational parameters for separation techniques

Ri and Rv studies demonstrate that reported assay values are not affected by small variations of “essential” operational parameters. In our design, Ri studies may be carried out using a one-factor-at-a-time approach for all essential parameters. Selection of those essential assay parameters can vary by method type. With the one-factor-at-a-time approach, it is important to establish “target expectations” for acceptable changes in output, which ensures that these studies do not repeat development work. The maximum allowable output change can be linked to target expectations for method precision, which are derived using the Horwitz equation (2,3,4,5).
Before MV and in conjunction with Qv, Rv robustness studies should be executed according to an experimental design (e.g., design-of-experiments, DOE) so the combined impact of essential parameters can be evaluated. In some cases, robustness studies are extended to some less important parameters. It is important then to add experiments performed at nominal conditions so method variability can be established and used as a “gold standard” against which the outcome of robustness experiments can be compared. Data analysis typically includes sophisticated tools for determining the statistical significance of each factor to method output, thereby guiding conclusions made about a method’s robustness.
Method QualificationTo meet current compliance expectations, an analytical method used to support GMP activities must be suitable for its intended use as documented by appropriate experimental work. Demonstration of method suitability can be divided into two sets of activities: qualification and validation. When methods are new, under development, or subject to process or method changes, this activity is often called qualification (6), whereas more formal confirmation of method suitability for commercial applications is called validation. Method qualification evaluates a method’s performance characteristics against meaningful target expectations. It is a critical development activity for establishing the suitability of a method for release of early to middlephase clinical materials but is not, to our knowledge, clearly defined in regulatory guidance. Such documents tend to focus on method validation.
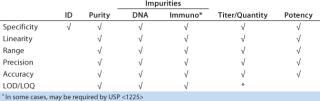
To reliably assess method performance (qualification), all necessary performance characteristics should be evaluated in carefully designed experiments. ICH Q2R1 specifies which performance characteristics should be evaluated for validation. But interpretation of the guidance for protein products is not straightforward. Table 2 details performance characteristics that should be assessed during qualification and subsequently during confirmatory validation experiments for such products. In this strategy, experimental design is the same for initial and late-stage method qualification (Qi and Qv). The only difference is the number of sample types to which the strategy is applied.
Table 2: Method performance characteristics evaluated during qualification and validation
To ensure the integrity of data provided on a certificate of analysis for clinical lots, Qi should be performed before a method’s implementation in quality laboratories. At early stages of clinical development, usually only one sample type requires qualification because in general both DS and DP for which specifications are established often have the same composition (formulation).
At later stages (but before process validation), Qv should be executed for all sample types that require routine GMP testing. This includes those listed on release and stability specifications (intermediates, DS, and DP), those associated with process controls and decision points, and those used during process validation (PV).
Method ValidationMV confirms performance characteristics demonstrated during MQ and demonstrates an analytical method’s suitability for commercial use. This effort should follow a preapproved protocol with clear and justifiable acceptance criteria. In the context of the analytical lifecycle, the key components of MV are its experimental design and acceptance criteria.
MD experimental design should mimic that of the qualification, and MV acceptance criteria should be linked to target expectations used in qualification experiments. In the absence of such rigor, validation experiments become mere exploratory research and run the risk of undermining the results of MQ.
Validation acceptance criteria should be aligned with target expectations from qualification. In most cases, acceptance criteria for validation are set based on the type of method and should not differ from target expectations. When target expectations are not met during a qualification study, the rationale for their reevaluation should be proposed in a qualification summary. Setting acceptance criteria for the precision of a method frequently causes confusion, anxiety, and inconsistency in practice. For validation, Horwitz’s predictions of relative standard deviation (RSD) for precision studies (detailed in Part 2) provide excellent guidance for setting acceptance criteria.
To prevent problems with subjective interpretation, validation acceptance criteria should include only objective parameters from the qualification. For example, during qualification scientists often expect that the residual from linear regression will show no bias (trend). Because the scientific community has not adopted a uniform measure of bias (which is frequently based on visual evaluation), it is inadvisable to include such requirements with validation acceptance criteria. However, this topic should be discussed in a validation report.
Validation studies are linked to process validation. They confirm the suitability of qualified methods for commercial application and are generally completed before release of validation lots. These studies should be executed for sample types that will be routinely tested in a GMP environment to make decisions about product disposition. Sample types include those listed on all release and stability specifications (intermediates, drug substance, and drug product) as well as those associated with process controls and decision points. We believe it is unnecessary to conduct method validation for sample types collected and tested only during process validation, for which qualification studies alone are appropriate.
Part Two of this article will address important scientific concepts and challenges associated with method qualification, validation, and remediation in more detail.