Earlier this year, the FDA issued its long-awaited process validation guidance document, which had been several years in development. It is well written and effectively articulates what many progressive companies have been thinking and doing for years. But many people in the industry are asking questions: How will it affect our process development programs? How will it affect the submissions and licensure of our products? And how will it aid in our commercial operations? Or will it have no effect?
Consider what the document says. In the simplest terms, it articulates four main messages. A strong process development program is essential — preferably using quality by design (QbD) — to eventually obtain expedited product approval and benefit from a relaxed regulatory position with the agency. Process validation (PV) is linked into that activity in verifying its conclusions: a set of team-based activities requiring many disciplines to work together to be effective. PV is value added and never stops. It requires a life-cycle approach with continuous verification and adjustment. (So continuous improvement is central to the activity.) By applying these principles, you will end up with a more reliable process that yields consistent products to meet your customers’ requirements, resulting in more satisfied customers. The overall take-home message is that PV is part of the drug development life cycle, that it never stops, and that it is a value-added activity. This can be described using what I call the “four Ds”:
- Design (build each process to a set of standards or requirements focusing on what a customer wants/needs)
- Demonstrate (by conducting experiments and validation to show that process and product meet design requirements)
- Document (the cornerstone of good work is recording it for posterity)
- Determine (ensure that results remain valid, or make changes as knowledge and experience grow — for continuous improvement).

A Little History
To determine what impact the document will have on all the industry’s questions and systems will require time. The answers rest in how companies use the guidance in process development, regulatory submissions, and operations. To fully appreciate its potential, however, step back and examine the status quo on PV’s role in product development — as it began and has evolved until now.
When it was first incorporated in the mid 1980s, PV was viewed as a new regulatory requirement to be performed sometime in phase 3 of the clinical development cycle. That was when processes were being locked down and companies hoped their products would be proven safe and efficacious. Whether going for a drug approval or biologics licensure, a company either submitted protocols and performed PV later (but before sale of the first lots) or submitted the completed PV reports with its submission. Once that was complete, you could put your reports on a shelf and forget about them unless you made radical changes to your product or process. This was not a value-added activity, but rather a check-the-box process.
Meanwhile, the FDA wanted all legacy products (those that had been approved before PV) to have their processes validated, too. This created a flurry of activity in the 1990s (and later) to retroactively validate those products. But again, the folders were then stored away and ignored. All that entailed a great amount of work with very little return on investment beyond keeping products on the market and keeping companies out of regulatory trouble.
That was the old paradigm. Over time, companies began to use PV activities to demonstrate ruggedness of their processes by choosing worse-case settings for critical parameters in three PV runs. This method brought value to the work involved, but it was impossible to examine all high and low conditions for all critical parameters in all combinations and permutations in just three PV exercises or runs. The work was labor intensive, yielding relatively little return on investment. However, the paradigm was changing.
Then came QbD, another FDA-driven concept. It was broadly placed into the “GMP in the 21st Century” initiative that came about because of several factors. The FDA was viewed by many detractors as very conservative and not amenable to change or to encouraging implementation of new technologies into processes and products — particularly for those that had already been approved. Further, the agency was under pressure of increasing workloads with a noncommensurate increase in resources. It was forced to do what industry has always done: more with less. Faced with that, the agency attempted to create new visions whereby industry investment into creating better interactions with regulators regarding process and product understanding might allow the FDA to relax its oversight somewhat. That would allow it to slacken the strings of control and give industry more opportunity to control its own destiny.
How QbD Works
The principles behind QbD are straightforward. If companies do a better and more systematic job of process development, then they will understand their processes and capabilities better. In turn, that will lead to more rugged processes that are more successful (and economically more rewarding). Manufacturers will understand what attributes of their products are critical to meet customer needs. And they will learn what factors contribute to process outcomes and how to control those more effectively. They would also gain understanding process inputs (raw materials) affect those outcomes. All of this will link inputs, process controls, and output (Figure 1).
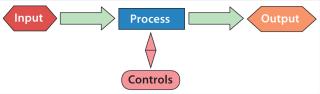
But that’s not all. Companies must take the results of validation studies and package them to convince the FDA that they understand their processes and how to control the outcomes. If you are successful, you will get regulatory relief. You might even avoid having a preapproval inspection (PAI) and be allowed to operate within a very wide “design space” with confidence regarding the outcome — even outside your clinical experience — without very extensive regulatory approval for changes. Of course, change control is required outside the operating range but within the design space — but that would be without regulatory oversight (Figure 2).
![]() rc=”http://lne.e92.mwp.accessdomain.com/wp-content/uploads/bpi-content/BPI_A_110910AR05_O__168935b.jpg” refid=”168935″ align=”left” style=”background-color:#f7f7f7;margin-bottom:5px; margin-right:5px; margin-left:5px;margin-top:5px;cursor:pointer;” >
rc=”http://lne.e92.mwp.accessdomain.com/wp-content/uploads/bpi-content/BPI_A_110910AR05_O__168935b.jpg” refid=”168935″ align=”left” style=”background-color:#f7f7f7;margin-bottom:5px; margin-right:5px; margin-left:5px;margin-top:5px;cursor:pointer;” >
To date, the best illustrations of QbD success are Pfizer’s Chantix and Selzentry products. The company had no PAI in the first case and was allowed to operate within its design space (using only internal change control) in the second. To date, I am aware of no equivalent successes in the biologics arena — yet. But Genentech (a member of the Roche group) has been very visible in leading the charge.
ICH Q8, Q9, and Q10 (and possibly Q11 when it is issued) admirably describe the elements of QbD by incorporating development principles, risk management, and progressive quality principles reminiscent of how the International Standards Organization (ISO) works for devices. A review of these documents will help you understand the major elements and nuances, but here are some key principles. In a system or product that has undergone QbD, the following elements are recognized:
- A product is designed to meet patient needs and performance requirements defined by its critical quality attributes (CQAs). In simple terms, if we make a product that meets its CQAs, then it works each and every time it is used as prescribed.
- To accomplish that, the production process is designed to consistently meet and attain a product’s CQAs. They may include specifications and other requirements not covered by conventional specifications.
- The impacts of raw materials and process parameters are well understood, as are their effects on outcomes (CQAs). Those with little impact are as well understood as those with major impacts.
- Critical sources of variability are thus identified and controlled by appropriate strategies. So you know what to control, and you control it to ensure that your CQAs are attained.
- The process is continually monitored, evaluated, and updated to assure consistent quality over time. This represents a state of continuous improvement.
With all that integrated into a development cycle, a company moves from operating reactively (obsessed with simply compliance for compliance’s sake) to proactively operating under continuous improvement. Experience and knowledge regarding its manufacturing processes do not stop in the development phase, but rather grows through this continuous improvement cycle. Ever-expanding knowledge and experience increases confidence in both product and process, thus giving a company the tools to improve them.
The development cycle defines a design space that does not stay static but is forever being refined. It is the area within which a company can operate its process with predictable outcomes to make acceptable products every time. This space is greater and broader than the operating ranges for routine operations. A company that presents this evidence to the FDA should be allowed to operate without restraint if it stays within this “space.” ICH Q8 describes in detail the elements for an effective pharmaceutical development program (1).
Quality Risk Management: Cost-effectively defining a design space requires introducing a further element: quality risk management (QRM), which is detailed in ICH Q9 (2). With multiple process parameters and raw materials inputs, a company faces thousands of potential experiments or questions to answer. Apply the principles of risk analysis and directed efficient experimentation (e.g., using design of experiments, DoE), then carefully analyze the impact on outcomes of raw materials and process parameters and controls. So you can prioritize a complex array of potential experiments and choose only those that will yield the necessary answers for defining your design space. Less effort is required to further explore parameters and inputs demonstrated to have little or no impact, and limited resources can be focused on those with significant impact.
To move from a compliance-obsessed reactive state to a process-knowledgeable proactive state requires a company to operate quality systems differently. You must still follow good manufacturing practice (GMP), of course, while incorporating other elements. ICH Q10 describes these elements in detail (3). Companies familiar with ISO processes will immediately recognize these as key tenets in that philosophy, as well:
- Management must actively ensure that quality systems are incorporated effectively into operations and champion continuous improvement for operations, products, and processes.
- Change control becomes a cornerstone process to help a company embrace the concept of product (and process) history files. Each and every change contemplated should move a product closer to customer requirements. This drives the manufacturing process into a more predictable outcome, ever increasing its ability to attain a product’s specific CQAs.
- Nothing remains constant, and everyone actively pursues continuous improvement, which becomes the driver for increasing knowledge after a product, process, and design space are developed.
Although Q11 is not yet complete, it will focus on much of what is described in Q8 while emphasizing that the principles of product development apply equally well to active pharmaceutical ingredients (APIs), bulk drug substances (BDSs), and drug products. This is well understood in the biologics industry but perhaps not so well appreciated in the pharmaceutical world at large.
The New PV Paradigm
With QbD, the front end of the PV cycle is clearly delineated. The new guidance provides a framework for phases 2 and 3 in this life cycle as well. It describes how PV activity links into process development and includes the concept of process monitoring and adjustment (elements of continuous monitoring and improvement).
With this new paradigm emerging, some people ask, do we stop validating processes as we have been doing so? Or must we rethink the mechanics? Inevitably, the answer is both yes and no. Many principles of PV still stand, but the reasoning (and hence the execution) may very well change. Clearly, the execution of classic equipment and facility qualification and method validations will not go away. But when you design and execute your qualification program, think of the end result in your program more than ever before. Qualification efforts will need to meet the needs of both product and process:
- Design qualification (DQ) — what exactly will we require the equipment or facility to do? What are our requirements?)
- Installation qualification (IQ) — is everything there and connected correctly?
- Operation qualification (OQ) — when you turn it on, does it function?
- Performance qualification (PQ) — does it reproducibly do what it is supposed to?
With those activities complete, you’re ready to begin the elements of PV, assuming you have completed the development phase and defined your design space. Your PV program should encompass all elements “from vial to vial” for biological products and further for drug products, including distribution systems. Roles and responsibilities must be defined because this is a team activity that requires the input of several disciplines. Typically, it should include at least members from production (the process owner), process development (technical experts), validation (the administrator and designer of a PV program), QC (sample testers), and QA (for oversig
ht and approval) groups.
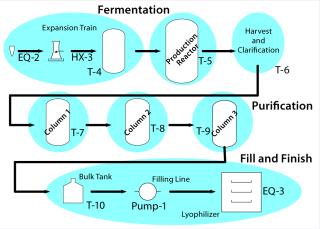
A Master Plan: You may eventually demonstrate your whole process — from vial to vial — but it helps to split the process into distinct unit operations with their own inputs (raw materials), control parameters, acceptance criteria, and output requirements. Figure 3 suggests how a biological process might be divided.
The examples below illustrate how to set up a validation program for specific unit operations. This incorporates familiar elements: validation master plans, protocols, and reports. Use what you have developed and are accustomed to using. Because biological product processes are highly complex, a master plan is a critical element to tie together each unit operation with its demonstration. As you develop each protocol for each unit operation, consider the following points in planning your activities (Figure 4): the unit operation itself, raw material inputs, process controls, and product attributes.
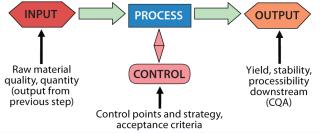
The unit operation: What is its purpose? Why is it configured the way it is? What is the role of each part? What is not accomplished in this operation that might affect steps downstream? All of those are defined by your QbD program.
Raw material inputs: What are they, what quality, and how much of each material is used? Which ones play significant roles in process outcomes? Which are otherwise benign?
Process controls: Which parameters must be controlled for product quality, and which for business results? What are acceptable ranges for each parameter? Do you know how to control them effectively?
Product attributes: What do you want as an end product for a given step? How does it tie into the next unit operation? One step’s output is a raw material for the next step. How does each one fit into a final product?
For example, consider the fermentor or cell culture production step for a drug substance and the final filling step for a drug product. Table 1 lists supporting details.
Looking Ahead
Part 2 will conclude this article with an upstream and a downstream example of applying these principles. It will also go into more detail about the postapproval phase as well as the implications of the new paradigm.
Author Details
Peter H. Calcott, PhD, is president of Calcott Consulting, 931 Mendocino Avenue, Berkeley, CA 94707; 1-510-585-8256;peterc@calcott-consulting.com; www.calcott-consulting.com.
REFERENCES