Currently the biopharmaceutical industry is transitioning to a new business model of production efficiency through implementing operational excellence (Op Ex). Borrowing from such principles as “lean manufacturing” and “Six Sigma” (6σ), and incorporating quality by design (QbD) (1), Op Ex is being applied through the implementation of such advanced enabling concepts and technologies as quality risk management (QRM) (2), process analytical technology (PAT) (3), and systems biology (SB) (4).
Some people see a conflict here: This paradigm shift is occurring amid ever-increasing product development costs, looming biogenerics and biosimiliars, and shortened product life-cycles. On the contrary, however, those very burdens contribute to the industry’s increasing need for innovation and efficiency, whether that be in product and process development (including control strategies), manufacturing, or quality assurance. And that is precisely what the FDA intended to address with its guidances on PAT and the quality systems approach to pharmaceutical CGMP regulations (5,–6).
Anticipated difficulties in applying QbD and PAT principles in biopharmaceutical production may have been overstated. Despite the inherent complexity of therapeutic proteins, manufacturers historically placed significant emphasis on product and process characterization, which has led them to better understand their manufacturing processes and the inherent sources and levels of variability therein (7).

Formulating the Value Proposition: Op Ex, QRM, PAT-enabled QbD, and SB can appear a priori as respectively representing disparate business, regulatory, or scientific drivers. But upon closer evaluation, their biopharmaceutical applications in fact not only share common objectives, but also promise quite a number of similar gains (Table 1). Implementation of the PAT guidance and the QbD and QRM guidelines may be voluntary, but the adoption of new quality concepts in process development, enhanced process control strategies, or risk management approaches may not be. Therefore, perceived value propositions and actual implementations may differ significantly between individual manufacturers. However, these transitional concepts, especially when combined, promise substantial gains that would behoove manufacturers to set out on their respective journey to capture them.
Table 1: How the goals and expected performance gains of operational excellence are achieved by PAT-enabled QbD/QRM concepts and systems biology approaches. APC = advanced process control; CPP = critical process parameter; CQA = critical quality attribute; SPC = statistical process control

Qualifying, classifying, and reducing variability in manufacturing processes has been a key theme in, for example, processes designed under the 6σ philosophy using statistical process control (SPC) as a principal enabling tool. In fact, this is a universal goal shared by Op Ex, QbD, and PAT alike. One useful way to categorize variations is to consider their origin as either common or special causes.
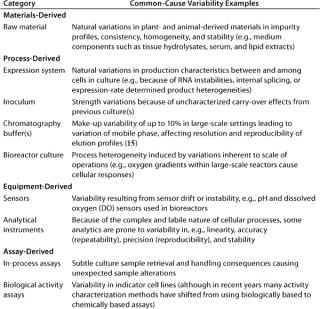
Common-cause variability (also known as inherent variability) describes random error or bias in a performance measure that produces an endemic deviation from the expected value in a process or its measurement. Such variability is inherent to each system as it exists, and this is predictable (probabilistically) and definable. It is the reason most operating parameters cannot be defined by single values, but rather by a statististically determined range around a mean value that spans multiple manufacturing lots. If a process displays only common-cause variability, it is traditionally referred to as being in statistical control. Only through improvements in the underlying design of the process itself can this zone of acceptable performance (within its upper and lower control limits) be reduced or refined. Table 2 lists examples of what today can be considered common-cause variabilities specific to a cell-culture–based biopharmaceutical production process.
Table 2: Principal categories and examples of common-cause variability specific to cell culture based biopharmaceutical manufacturing processes
Special-cause variability, on the other hand, is nonrandom, transient, and often assignable in nature. By definition this type of variability is not inherent, predictable, or definable. Such out-of-statistical control occurrences are addressed using a problem-solving approach to determine their root causes, local remedies, and/or methods of elimination.
Common- and special-cause variability are empirically assigned categories, usually based on history and current understanding of a process that has nothing to do with the essential nature of their root causes.
For example, a sensing system containing an unrecognized defect that produces chronic noise (at an acceptable level) throughout process development could be categorized as demonstrating common-cause variability. Consequently, it could be addressed by setting control limits around a target value. However, had the same type of sensor performed significantly better throughout process development, a narrower range of limits might have been set.
Subsequently, if exactly the same defect occurred in one particular sensor of that type, then the system would be out of statistical control, which would thus reflect an incident of special-cause variability. As functional understanding of a system increases, variabilities previously categorized as common-cause, predictable, and unassignable may be recategorized as special cause by application of that increased understanding.
Each variance typically results not just from action at the current step, but rather represents an accumulation of variation from previous steps, including measurement errors. So an important question that must be answered in process characterization is how variation from different sources accumulates or propagates. That allows us to allocate the total process variability across an entire manufacturing process and then focus on those that contribute most to it. See “Process Robustness” in the online appendices for ways of reducing product variation that results from process in-put variability.
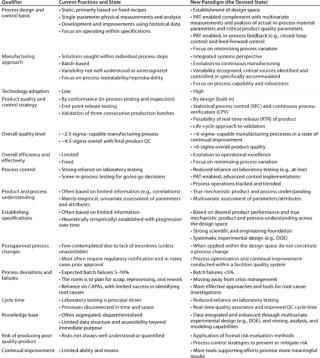
Quality Redefined: From Product- to Process-CentricIntegration of these new initiatives should lead to an evolution of biopharmaceutical product and process development and manufacturing practices from their current state to a new desired state (Table 3). This applies equally in both the development of new products and in the optimization of existing processes.
Table 3: Projected evolution of biopharma’s current practices and state to the desired state by implementing the initiatives of the new operating paradigm
The Current State: Regulatory authorities worldwide have undoubtedly contemplated process-centric rationales in their regulations, guidelines, and guidances for biopharmaceutical manufacturing and drug approval processes. However, having surely been challenged by the sheer number of biotech products and the resulting variety of processes, they have in practice followed a more pragmatic, product-centric regulatory approach. The reality is that quality determination of each end-product (e.g., drug substance and drug product) is often relegated to batch and release testing, which simply distinguish between conforming and nonconforming material.
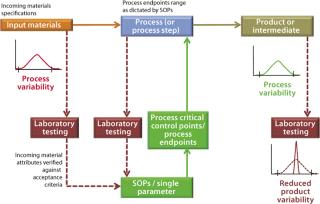
For example, Figure 1 outlines an abbreviated process control model for a biomanufacturing process by highlighting key elements of the current state. This can be compared directly with the PAT-enabled, desired-state process control model in Figure 2. The process control model selected for Figure 1 is admittedly the most rudimentary, yet still commonly encountered form, relying solely on laboratory testing and process standard operating procedures (SOPs) to establish end-product quality. As part of its overall quality control strategy, process control is predominantly established through
-
documented evidence (e.g., an executed batch record) of conformance to SOPs ensuring that manufacturing operational conditions are followed as dictated
-
insurance that raw material, in-process, and drug product quality characteristics fall within their established acceptance criteria by laboratory evaluation. Only for specific cases in which a manufacturing process can be validated for that(e.g., removal of a particular impurity) may testing actually be simplified. For example, validating a process for its capability to remove residual DNA or viruses (viral validation) precludes the necessity to test for the presence of residual DNA and viruses in the end product.
All batches manufactured must adhere to established process control procedures and acceptance criteria that have become the de facto regulatory commitment upon submittal of an application dossier. This rigidity in manufacturing operations can actually become a burden when original regulatory submittal documents (particularly the chemistry, manufacturing, and controls or CMC section) are based on limited process understanding. This is largely because subsequently contemplated process changes — not to mention those actually required in leveraging manufacturing experiences to ensure product quality — require regulatory notification and, in many cases, prior approval (8,9,10,11).
The overall outcome is quality by conformance, of which a major subset is quality by inspection, and a static manufacturing operation. Unfortunately, this commonly lessens incentive to implement postapproval process changes and limits opportunities for meaningful continual improvement.
Continual or Continuous Improvement
Some authors prefer the term continual improvement over continuous improvement. Although both are similar, they may invoke different meanings within a certain context. We chose to use the first term throughout these chapters for consistency.
Process Deviations and Failures: The complexity and (currently) static nature of biotech manufacturing processes can be exacerbated by often-limited initial product and process understanding. So it is not surprising t
hat pharmaceutical manufacturers expect deviations from established SOPs, process failures (such as out-of-specifications or OOS results), and even batch failures to occur. According to some analysts, these may occur at a rate as high as 5–10% (12). Consequently, the industry norm is to plan for scrap, rework, and reprocessing as appropriate.
OOS investigations require significant resources and may even scrutinize entire process steps (unit processing operations or UPOs) and procedures — but remarkably, they often fail to identify the root causes of OOS results. This can be in part attributable to a limited understanding of process variability and a lack of resources or ability to revisit the fundamental product and process design and control aspects that led to established specifications in the first place. It is difficult to determine what went wrong if you don’t know precisely what makes things right.
Addressing the root causes that lead to process failures may well be our best overall strategy to improve process efficiency and reverse the trend toward increasing manufacturing-related problems (e.g., recalls and disruption of manufacturing operations) currently witnessed in the pharmaceutical industry at large (13).
Overall Process Efficiency: Through data mining of existing batch records, industry analysis of average cycle times in actual manufacturing processes indicates that quality testing is discontinuous. This drives overall cycle times to such an extent that QC cycles can actually be longer than process cycles (14). In essence, some elements of pharmaceutical manufacturing processes are disconnected in time and space — rather than processes that could use in-line or at-line technologies for continuous process verification. Industry analysts estimate that for the pharmaceutical industry as a whole, the relationships between value-added and nonvalue-added product activities (as measured through direct time contributions in actual manufacturing operations) are 10–50%. Asset use is estimated at just 15% (12).
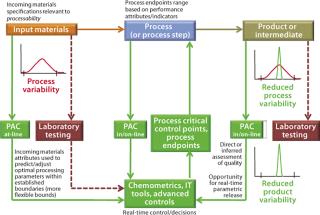
That contrasts with practices in other process-based industries, in which output product quality is maintained by adjusting key process conditions (e.g., on the basis of in-process measurements of product quality) to prevent or accommodate process variability. Such practice is referred to as in-process feedback or closed-loop control — as opposed to mere fixed recipes.
The Desired State: A PAT-enabled process control model (Figure 2) uses multiple process analytical chemistry (PAC) tools or analyzers for in-process measurements of raw material, in-process, and end-product quality attributes. Resulting data provide the input for advanced feed-back and feed-forward process control strategies that can be implemented using modern chemometrics tools. Such approaches include information technology (IT), bioinformatics, and the latest in control platforms such as statistical process control (SPC) and model predictive control (MPC).
Most manufacturers currently rely on many univariate in-process measurements of physical parameters (e.g., bioreactor pH, DO, temperature, agitator speed, and gas flow measurements) to help steer each process. However, such measurements usually are not directly correlated to in-process product quality attributes, although they may be able to imply some. Multivariate, real-time, closed-loop product quality control strategies for coping with process variability are not yet achievable. Such process control strategies are clearly the subject of the desired state, and they promise to increase overall process efficiencies.
The principles outlined here apply equally to process control models developed for entire manufacturing suboperations such as cell culture, clarification, purification, and fill–finish as well as stand-alone UPOs.
Process Innovation: Making a Business CaseA new risk-based QbD approach to innovation in biomanufacturing is most likely to vary in implementation from product to product, process to process, company to company, and even from facility to facility. But regardless of the tactical approaches taken, implementation success will require marrying a solid business case with strong science-and engineering-based process understanding by taking an integrated systems approach. Both process understanding (see Chapter 2) and process robustness are core concepts in the new regulatory framework, and they are the central underpinnings in steering process design and control activities.
Considering this new framework, the field of systems biology has fortuitously emerged at the right time. It promises to become a cornerstone for enhancing process understanding over several process steps.
Ultimately, if companies are to take full advantage of the new regulatory paradigm, their application of new technologies (16) and novel product and process design, monitoring, and control approaches to increase understanding and minimize variation in existing processes will fuel a business case for process innovation. These components form the core objectives of quality as well as productivity enhancement and improvement initiatives. Industry participants have an opportunity to apply them across all future process design and control activities.
Changes Pending: The Industry’s ReluctanceMany people in the industry have realized all along that quality by conformance, and release testing in particular, can neither predict nor improve product quality. Nevertheless, stakeholders are often reluctant to implement significant process changes to improve on in-process quality (e.g., by adopting new analytical technologies with in-process feedback control strategies). Reasons include the associated technical challenges, return on investment (ROI) issues, regulatory complexity, and scientific rigor required.
The industry is generally reluctant to introduce new technologies because of enduring mindsets from cultural and/or experiential heritage — and either principal regulatory, organizational, and scientific concerns as described in the “Different Perspectives” box. However, by recategorizing these concerns as challenges instead, we not only appreciate how comprehensive and interrelated they are, but we can also value the opportunities they represent.
From an engineering perspective, opportunities for improving efficiency and quality assurance through heightened focus on design and control, have either generally gone unrecognized or have failed to generate much enthusiasm (17). Consequently, development of novel bioprocess analytical technologies and instrumentation is often pursued only by small entrepreneurial companies and academia. Their pioneering spirit contrasts with the more incremental approach commonly exercised by established instrumentation vendors responding to the generally conservative approach their pharmaceutical clients have toward new technologies.
Despite several biopharmaceutical industry innovators, the adage If it isn’t broken, don’t fix it! has often emerged as a de facto paradigm for many companies because the regulatory and organizational burden of implementing changes appears to outweigh potential economical gains. The consequent process technology status-quo often encountered in the industry is probably not what regulators actually intended, but it happens often to be the outcome of a cautious, conservative approach to managing regulatory (and ultimately business) risk.
DIFFERENT PERSPECTIVES, DIFFERENT CONCERNS
Here are some examples of regulatory, organizational, and scientific concerns regarding the implementation of new technologies by biopharmaceutical manufacturers.
Regulato
ry Concerns
General concern of “raising the bar”
Real or perceived fear, justified or not, of delays in regulatory approval for new products due to many questions raised
Fundamental concerns about regulatory implications of having new manufacturing process information available (in particular by applying PAT) that wasn’t previously seen or known: It is generally accepted that closer scrutiny of processes may reveal variations in existing products that are missed by current monitoring and sampling techniques. If applied to already-approved products, that might in theory jeopardize their state of GMP compliance (18).
Uncertainty regarding how regulators will assess method suitability and validation — especially for those involving novel techniques (e.g., based on pattern recognition and chemometrics)
Organizational Concerns
Reluctance toward conducting extensive comparability studies, further characterization work on existing products, bioequivalence studies (especially those requiring clinical trials), and preparation of elaborate documentation submittals
Unwillingness or inability to allocate organizational resources and time to develop the above, especially for duplicate objectives (e.g., methodologies and controls) when the existing ones work
Uncertainty about economic incentives — reinforced by the general lack of a systems perspective in process design and control — and their direct investment costs (e.g., in equipment)
General feeling of a “time crunch” to achieve the highest level of science and engineering underlying comprehensive process development efforts
Scientific and Technological Concerns
Unfamiliarity with newly proposed technologies and their specific value in a given application
Absence of accepted scientific standards to ensure proper calibration, method validation, and operation of such technologies
Uncertainty regarding which technology to use because of a general lack of industry benchmarks and established practices
Challenges in aseptic sampling, achieving real-time on-line instrument settings, and validating instrumentation, as well as safety and data integration
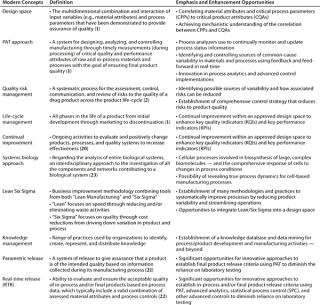
Although PAT has found a home under the QbD umbrella, many biopharmaceutical companies are still unsure whether all these modern concepts (Table 4) can be aligned beside pressing business needs to render manufacturing processes more efficient and cost effective. While acknowledging the promise of many specific benefits, many people pause at the more comprehensive question: “Will biomanufacturing under the new framework truly become more efficient and effective?”
Table 4: Some modern business and regulatory concepts useful in biopharmaceutical development (and establishing quality enhancement programs)
Anyone doubting the imperative of Op Ex, QbD, and QRM principles in ongoing biopharmaceutical development, however, may observe that ASTM International Committee E55 on Pharmaceutical Application of PAT has already approved several practices (19). In the past, each qualitative step in advancing efficiency or quality rigor has been met with initial skepticism and resistance. Yet we could not now imagine operations in a context without ISO, GLP, GMP, or GCP practices and quality management systems.