 High costs and long timelines for biopharmaceutical development are cause for reflecting on how best to allocate resources from the earliest discovery stage through critical go–no-go junctures. With inputs ranging from science, engineering, and economics, the coined term developability becomes the synthesis of answers to such questions as
High costs and long timelines for biopharmaceutical development are cause for reflecting on how best to allocate resources from the earliest discovery stage through critical go–no-go junctures. With inputs ranging from science, engineering, and economics, the coined term developability becomes the synthesis of answers to such questions as
- How well does the target represent a disease state?
- Does manipulating that state bring about improvement?
- Does the molecule behave as expected in living systems?
- What can be done about the emergence of independent safety, toxicology, and/or
immunogenicity warning signs? - Can the molecule be made at scale for the right price?
Attrition of therapeutic candidates during clinical development is the major factor in high development costs. Dollar and year figures vary too widely to quote any single source. And related statistics are based on averages, incorporating resources devoted to molecules that fail. Gaining control over the timing of failure through developability assessment becomes critical.
Developability assessment teams are multidisciplinary by necessity. They are charged
with evaluating potential drug candidates to allow pharmacologically effective new biological entities with favorable toxicity/immunogenicity and efficacy profiles to proceed through development. This presumably decreases late-stage attrition.
“Although still grounded in safety, efficacy, and pharmacokinetic/pharmacodynamic (PK/PD) considerations,” explains BPI editorial advisor Hazel Aranha, viral clearance and safety manager at Catalent Pharma Solutions (Morrisville, NC), “early stage developability assessment considerations may be affected by such nonscientific factors as stock market and shareholder expectations.” Today’s focus is on cost containment and operational excellence facilitated by multiple advanced upstream and downstream initiatives, including generation of genetically engineered, high-yield expression systems and disposable manufacturing that eliminate the need for traditional brick-and-mortar, greenfield, single-product facilities. Developability as a discipline is better established for small-molecule drugs than for biopharmaceuticals. “The most common application of the term developability for biologics involves assessment of potential manufacturing risks such as product stability or aggregation,” says Jesús Zurdo, senior director of strategic innovation at Lonza Pharma and Biotech (Cambridge, UK). “However, we and others have been advocating for a more holistic approach to developability that incorporates other factors that could be critical for the success of a new therapeutic candidate.”
Those include aspects of safety (e.g., immunogenicity, adverse reactions), formulation (affecting product delivery, but also potentially the cost of treatment), pharmacology (particularly for nonantibody therapeutics) and more elaborate aspects of efficacy that go beyond target binding. “All these require developing new methodologies for testing and assessing risk,” Zurdo says.
Early Stage Developability
Three Levels to Consider: Until recently, early drug discovery focused exclusively on a molecule’s PK and PD, with efficacy/toxicology assessed through in vitro and in vivo models. Increasingly, however, developers are evaluating intrinsic properties that influence the technical development of drug candidates early in R&D. “This developability assessment serves to minimize the risk of facing major challenges during drug substance production and drug product formulation,” says Christoph Freiberg, senior scientific consultant at Genedata (Basel, Switzerland).
Genedata’s services include identification of predictive developability parameters based on data collected from multiple R&D projects. Such efforts are facilitated by the company’s biologics platform, which collects molecule, sample, and analytical data during a product candidate’s life cycle, then integrates all that information across all projects within an organization.
Genedata identifies three levels of early developability assessment. First, during drugcandidate screening, early elucidation of a molecule’s primary structure is typically accompanied by an in silico analysis of potential liabilities. Common problems include aggregation propensity, formulation instability, and reduced pharmacological activity. The company combines in silico analysis and engineering tools with the management of screened and subsequently engineered molecules’ gene sequence and associated analytical and assay data. Those results help to define subsequent screening strategies and early engineering interventions that may improve developability.
The second level involves expression and purification — manufacturing risk, as addressed toward the end of this report. The third tier involves assessment of properties affecting a protein’s robustness. Depending on the number of molecules tested, limited or extended sets of assays are applied to address their biophysical and biochemical properties. Such assays address stability (pH, freeze–thaw, and high- and low-temperature stability tests over limited time frames), degradation (forced by extreme pH values, light, and oxidation), viscosity, thermal stability, solubility, and glycosylation characterization. Because such characteristics relate to immunogenicity, some groups also include in vitro and/or in vivo surrogate assays for assessing immunogenicity as part of their developability assessment. Doing so enables in-depth developability assessment during the transition from research to development.
“Developability assessments are increasingly performed earlier in discovery,” Freiberg says, “for example during screening, which means dealing with more molecular entities and more heterogenous data.”
| Biologicals Are Different by Christoph FreibergIn general, both small-molecule drugs and biological molecules interfere with physiological processes: On one hand, they show therapeutic efficacy; on the other hand, they can cause unwanted side effects. Thus the major focus of early toxicity studies remains similar for both molecule types. Developers need to clarify whether side effects might be caused by each drug candidate’s mechanism of action.But toxicity studies are different for classical drugs and biopharmaceuticals. Biologicals are easily detected in histological sections, so analysts can perform tissue distribution studies on them early in development. According to safety guidelines from the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, these studies are “essential in providing information on distribution and accumulation of the compound and/or metabolites, especially in relation to potential sites of action; this information may be useful for designing toxicology and pharmacology studies and for interpreting the results of these experiments” (1).Early immunogenicity assessment is unique for biologicals. The safety consequences of immunogenicity can vary widely and often are unpredictable in patients who are given a biologic. Most biopharmaceuticals induce immune responses, which in many cases are clinically irrelevant. For example, the appearance of antidrug antibodies (ADAs) in most patients is nonsignificant. In some cases, however, ADAs can reduce the therapeutic efficacy or generate side effects ranging from minor hypersensitivity reactions to anaphylaxis through cytokine release syndrome.Detection of ADAs is one early parameter to be determined, although such findings in preclinical animal species aren’t always truly predictive. Developers also perform in silico analyses and computer modeling to predict primary structure-related immunogenicity. In vitro assays help them study responses of immune system cells. All these tests are artificial, however, which is why animal studies (especially using nonhuman primates) are indispensable.Immunogenicity is monitored in patients by measurement of ADAs in their blood. Other parameters that indicate elevated immune responses also might be monitored (cytokines, immune cells, and so on).Organ Toxicity Organ toxicity relates to a drug’s adverse effect on specific bodily organs. For example, skin rashes or other reactions do not necessarily have to do with immunogenicity of a given drug. Many cancer treatments — whether small molecules or biologicals — have dermatological side effects due to their modes of action.Organ toxicity is studied through measurement of physiological blood parameters and histological studies. For example, detection of certain enzymes in blood samples indicates damage to liver, kidneys, or other organs.Toxic effects in organs where a biological entity can concentrate are monitored using methods similar to those in classical organ-toxicity studies. Immunotoxic events can be triggered because certain biologicals (e.g., therapeutic antibodies) can trigger local immune responses through which bound target cells are destroyed by immune cells or immune factors. Examples include “antibody-dependent cell-mediated cytotoxicity” and/or “complement-dependent cytotoxicity” cascades.Reference 1 ICH S3B: Pharmacokinetics: Guidance for Repeated Dose Tissue Distribution Studies. US Fed. Reg. 60(40) 1995: 11274–11275. Christoph Freiberg is a senior scientific consultant at Genedata in Basel, Switzerland; www.genedata.com. |
Zurdo adds it is never too early to think of developability. “I would argue for considering
developability from the moment a new product concept is conceived.” He holds that
comprehensive developability involves not only the desired target, but also requirements for successful commercialization — e.g., required half-life, dosage form, administration route, and cost structure for treatment (including manufacturing, clinical administration, and even reimbursement).
He offers the example of a new development stage drug intended to treat an autoimmune condition and thus requiring chronic dosage. Besides elements such as mechanism of action, target, and format, success in the marketplace requires a formulation that is suitable for patient self-administration, with adequate guarantees against immunogenicity risk.
“These two factors (formulation and immunogenicity risk) are usually not well covered during early stages of biopharmaceutical discovery, or even development.” Formulation development typically occurs later on, sometimes even after phase 2. At that stage, fixing unexpected issues with stability or viscosity at the required dosage concentration cost time and money. “But you might also fail, and at a very high cost,” Zurdo warns. “This is not a mere possibility; it really happens. Wouldn’t it have been better to design and select from the beginning molecules that comply with formulation and delivery requirements? Surely, development would be considerably less bumpy and dramatic.”
Addressing Fundamental Developability Risks: At the Integrated Biologics Profiling (IBP) unit in biologics R&D at Novartis Pharma AG (Basel, Switzerland), very early stage developability assessment on monoclonal antibody (MAb) candidates is guided by the need to address fundamental risks of aggregation, hydrophobicity, conformational and interface stability, and inappropriate isoelectric point. The team establishes high-throughput analytic protocols that are suitable for evaluating a large number of candidate MAbs.
To enable rapid processing times, product candidates are produced in a transient human embryonic kidney (HEK) cell expression system, and each protein molecule is tested for the parameters mentioned above.
“After selecting up to four molecules that fulfill the best combination of biological activity, biophysical properties, sequence, and epitope diversity, we initiate the full profiling phase,” explains one senior member of the IBP team. Profiling entails small-scale production that is somewhat representative of upstream and downstream clinical manufacturing processes, with detailed analysis of physicochemical properties, stability assessment in selected formulations under representative stress conditions, and in vivo fitness.
“Final developability assessment provides a very detailed characterization of a molecule with respect to aggregation at, for example, different concentrations,” the team leader continues, “but also viscosity, solubility, clipping, or any other posttranslational modifications. This is when we perform specific in-vivo fitness tests such as Fc-receptor binding studies.” For nonantibody therapeutic proteins, such steps are adapted to the specific needs of each molecule.
To ensure that only viable candidates enter the next round of optimization, the IBP unit conducts very early stage developability assessment through screening of phage-display libraries. Full assessment occurs as soon as up to four lead candidates become available, one of which becomes a clinical candidate.
“With significant challenges, such as loss of potency because of unknown posttranslational modifications (a very rare event) or inappropriate biological readouts during dose-range–finding (DRF) and PK/PD studies, we will adapt R&D plans accordingly,” the team leader adds. “In our experience with developability assessment for standard antibodies, extreme cases of technical issues can be considered as an exception.”
Stopping the Show-Stoppers: Immunogenicity signals become tmost likely show-stopper during early development. Afterward, solubility and stability are significant concerns. All may be overcome to some degree through conventional clone selection or process-related techniques.
Additionally, regulators are increasingly concerned about stable nonstandard protein
conformations. These are often benign, but they may be indicators of a tendency toward immunogenicity or aggregation. Identifying the genetic constructs responsible for unfavorable protein conformation can assist in assessing early stage developability before immunogenicity and manufacturability assessment.
Another early stage red flag is the presence of a free sulfhydryl group, which presents a potential site of undesirable disulfide crosslinking. Few therapeutic proteins carry these moieties, but for those that do, developers might need to protect them in some way during downstream processing and formulation, perhaps by providing a chemically reducing environment. In some cases, a cysteine might be protected by protein folding (as in human serum albumin). Developers should assess very early in development whether a cysteine is required for activity. If not, perhaps an alternative amino acid with superior manufacturing properties could replace it.
Similarly the presence of certain posttranslational modifications — particularly
glycosylation — can send developers back to clone picking or even cell-line selection. Glycosylation profoundly affects biological activity, blood clearance, and antigenicity. For example, a lack of N-glycolyl neuraminic acid (NGNA) is a hallmark of human-like sialylation. Humans spontaneously raise antibodies to glycoproteins containing those structures. On the other hand, a complete lack of fucosylation is associated with significantly higher effector function that can enhance anticancer activity. Fucose-free glycoproteins are considered “next-generation” therapeutics.
Work-around strategies for mitigating unfavorable immunogenicity, solubility, and
stability depend on when such problems arise. That is why early developability assessment supports more straightforward problem solving. Reengineering lead candidates (or selecting alternatives) are options that have helped companies overcome developability challenges, according to Zurdo. For immunogenicity, formulation work might overcome factors beyond primary sequence (e.g. degradation, chemical modification, or aggregation). To address stability issues, he suggests investigating alterations in process parameters from cell culture to primary recovery, including specific process conditions, buffers, or even raw materials. “Having said this, things proceed more easily if a product is intrinsically stable, which is why early design and assessment methodologies pay
dividends.”
When faced with unfavorable immunogenicity, solubility, or stability, often your best course is not to seek work-arounds, but rather to minimize the impact of negatives. “Immunogenicity is difficult to determine in advance,” notes consultant and BPI editorial advisor Scott Wheelwright of Complya Asia (Shanghai, China). “Solubility and stability require development to evaluate alternatives and come up with a workable result.”
When redesigning a protein’s amino-acid sequence is out of the question, developers should focus on optimizing nucleotide sequences in the gene construct related to translation. “Different sequence features in both coding and noncoding regions of the constructs can improve protein processing,” says Daniel Ivansson, GE Healthcare staff research engineer for strategic technologies, “and hence reduce aggregation or improve solubility, secretion, expression, or other quality attributes.”
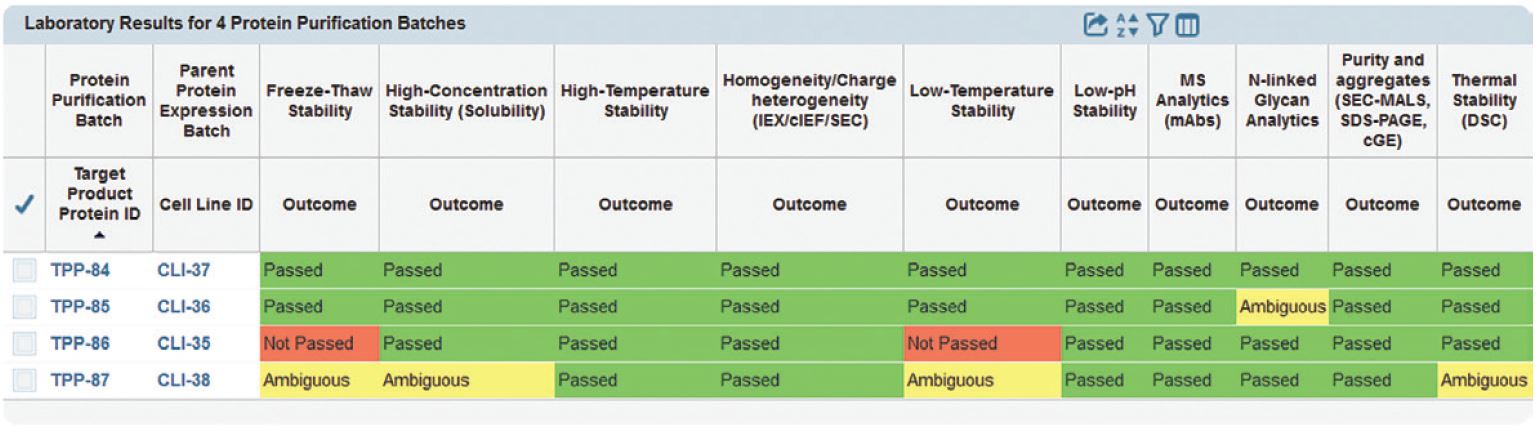
| Screenshot from a Genedata Developability Assessment
In this “traffic-light system,” individually reported quantitative and qualitative results from multiple analytical methods (often from different laboratories) are individually reported to the Genedata Biologics system and seamlessly compiled into one centralized, integrated view. The output below summarizes results of each test for purified samples (“Protein Purification Batches”) of individual biotherapeutic candidates (“Target Product Proteins,” first column). Proteins were expressed using distinct stable cell lines (essential for the “Parent Protein Expression Batch,” second column). Color-coded outcome fields indicate whether results do (green) or do not (red) meet developability criteria — or are ambiguous (yellow). |
Similar strategies may be adopted for difficult to-express proteins that would otherwise be
undevelopable. When those sophisticated strategies are unavailable, developers can fall back on advanced clone-selection methods to achieve desirable quality and expression.
| Manipulating Noncoding Regions by Daniel IvanssonProtein sequence is the dominant factor dictating expressability and protein conformation. However, effective translation of a recombinant mRNA into a functional protein (and the protein’s subsequent secretion) are controlled by a large number of interconnected cellular processes. An increasing body of research shows a link between proper folding of a defined protein sequence and the context in which that sequence is produced. Because folding is a cotranslational process, all factors affecting translation can influence the resulting protein’s conformation within the conformation free-energy landscape (which is dictated by the defined protein sequence).In turn, final conformation has a direct link to aggregation propensity. Conformations displaying hydrophobic patches are “sticky” and hence prone to aggregation. That reduces solubility/stability and target-binding specificity while increasing viscosity in high-concentration formulations. Besides complicating cost-effective protein production, all these factors present concerns for drug safety because they can affect immunogenicity and off-target effects.Coding and Non-Coding mRNA A recombinant mRNA sequence contains both coding and noncoding segments that dictate cellular processing. Amino-acid sequence obviously constrains the coding sequence, but even a protein as small as 30 kDa can be encoded by 10100 different nucleotide sequences, all yielding the same amino-acid sequence.Different nucleotide sequences are not processed equally by a production cell. An absence of splice-acceptor sites, miRNA binding sites, and/or restriction enzyme sites is an important factor influencing mRNA abundance. Many other factors can affect the translation of recombinant mRNA, leading to protein elongation, folding, and secretion in host cells. Although no consensus exists regarding precise mechanisms for how nucleotide sequence affects protein quantity and quality — and to what extent this occurs — the nucleotide sequence is clearly much more important than previously thought. Its effect on protein quality (including final conformation) is probably significant. This is evidenced by statements of FDA-associated researchers who say that future filings will probably need to include not only amino-acid sequences, but also nucleotide sequences of protein therapeutics (1).Synthetic Biology Opportunities Inefficiencies in coding or noncoding nucleotide sequences cannot be addressed fully through traditional strategies of clone selection, culture-media customization, and process development. GE Healthcare’s Life Sciences Business takes a holistic approach by offering tailored total upstream solutions. The company uses synthetic biology to design and develop an expression toolbox with a preoptimized set of 5’-leader sequences and signal peptides. That maximizes translational initiation, prevents negative consequences of alternative initiation, and provides for highly efficient secretion.A poorly behaved sequence around the intended initiation codon of the protein-coding sequence will either reduce efficiency of translational initiation (e.g., relating to secondary structure), create competing initiation sites in/out of frame, or a combination of both effects. At a high overall protein production load (including efficient recruitment of ribosomal particles), low-efficiency initiation and/or competing initiation can in turn seriously affect cellular behavior and the quality of protein processing. In my opinion, sequence elements and coding principles found in naturally evolved organisms are not a preferred way to address the above challenges. Sequences have evolved during conditions with evolutionary driving forces other than optimal efficiency in the processing of one type of protein class. The biopharmaceutical industry needs to take advantage of opportunities that synthetic biology offers in terms of writing nucleotide sequences. Upstream process engineers can learn the principles for how different sequences are processed during true bioprocess conditions in an intended host. DNA 2.0 in Menlo Park, CA, is one strong player that has adopted this philosophy. Reference Daniel Ivansson is a staff research engineer in strategic technologies for GE Healthcare’s Life Sciences Business. |
Process Development and Scale-Up
Assessment of manufacturability and process scale-up take the foreground as molecules progress from phase 1. Ruta Waghmare, worldwide director of emerging biotechnology at EMD Millipore (Billerica, MA) recommends that preliminary process development include recovery and purification unit operations that are not fully optimized from a sizing perspective. “The goal at the preclinical stage is to get acceptable purity levels for a product to get to the next stage of development.”
During phase 1–2, companies often further investigate each molecule’s process operating conditions using design of experiments (DoE) or take an even deeper dive into quality by design (QbD). “From a perspective of facilities and systems needs,” Waghmare notes, “the considerations for manufacturing from phase 1 onward should include project timelines, process economics, manufacturing flexibility, personnel expertise, facility infrastructure and available equipment, and batch volumes.”
Additional considerations during phase 2–3 process development include lot-to-lot variability of raw materials and consumables as well as batch-to-batch process variability. These factors provide fuller understanding of each unit operation and of the process performance as a whole. Both are critical for robust scale-up and manufacturing.
“For cases in which those deeper approaches are not feasible,” Waghmere continues, “appropriate safety factors can be incorporated for all unit operations to minimize the risk for process deviations and increase process robustness. Technology transfer and method transfer have to be considered as well, in even more formalized fashion if a contract manufacturing organization (CMO) is involved. Validated throughput during certain process steps can significantly affect the manufacturing footprint.”
Overall, each step in recovery and purification of biologics must be optimized based on process requirements and molecular characteristics to ensure robust, stable, and scalable production processes. “In the frenzy of target validation and process design,” says Hazel Aranha, “scale-up is sometimes addressed on an ad hoc basis.” Developers may need to reconsider critical process parameters defined during process development in light of changes in equipment scale, raw materials, or processes. “Possessing a thorough understanding of defined, parameter ranges — with data to back it up — is critical to scale-up and process validation.”
Analytics in Process Development: Analytical methods are key elements in both discovery and development of innovative biologics, although the two usually involve different technologies. “I do not normally see them as gating items in the decisions to bring product candidates through the pipeline,” says BPI editorial advisor Nadine Ritter, president and analytical adviser at Global Biotech Experts (Germantown, MD). Selecting biosimilar product candidates, however, is first driven entirely by analytics. “Without superior analytical methods to characterize the structure and function of candidate biosimilars, followed by head-to-head analytical experiments establishing an acceptable degree of biomolecular similarity to a reference licensed biotech product, a sponsor may not get regulatory approval to proceed with the necessary clinical trials.”
For both originator and biosimilar products, process development is the most direct operational factor affecting developability. Sponsors must end up with an optimized manufacturing process capable of consistently generating the drug substance in high yield, with minimal process residuals and specified quality attributes, preferably at manageable costs. And with both types of biologics, Ritter says, analytics are vital for characterizing process design space (for QbD) and establishing comparability after process changes. “But for biosimilars, the major analytical studies are significantly more front-loaded in project chemistry, manufacturing, and control (CMC) timelines. And they can be true project gating items for ongoing product development milestones.”
Freiberg observes that “a big challenge is the diversity of data and assay and analytical techniques, and the number of laboratories involved in generating such a panel of developability profiles.” Data also must be available on demand in a centralized repository to support transparent decision-making. “A centralized platform flexibly handles diverse analytical and assay data and allows single-point data access and generation of full developability profile reports.”
In addition, data collected during development must be communicated back to researchers so that
they can learn from experiences outside their group. Data-management systems that capture and structure key information from both research and development facilitate reengineering molecules when necessary.
| Defining Biosimilarity
How we define biosimilarity depends entirely on the type of protein, expression system, target indication, and other factors — and how they relate to clinically meaningful attributes of a protein therapeutic. Any of those elements can potentially affect product safety and efficacy. “You don’t know what you don’t know until you attempt to measure it,” says Nadine Ritter of Global Biotech Experts. An original drug’s sponsor couldn’t use modern, high-resolution analytical tools to characterize its product decades ago because those tools did not yet exist. But biosimilar developers must start out using a full slate of physiochemical and functional methods to assess biomolecular similarity for elements that even originators may not have measured or considered. Ritter says that is because each original product went through extensive clinical trials to qualify the ranges of all of its characteristics (even those the innovator could not measure analytically) in support of safety and efficacy. “Once a biopharmaceutical is approved,” she explains, “the product should remain consistent within clinical ranges as long as the manufacturing process remains in control.” When a new version of that product (a potential biosimilar) seeks to leverage the clinical success of the original, its developer should assess as many biomolecular features as possible for analytical similarity. The more parameters can be shown to align, the stronger the case will be for biosimilarity. “It is thus to the advantage of a biosimilar developer to conduct an extensive analytical interrogation of biomolecular characteristics. If the product then exhibits characteristics that are dissimilar from the original, then potential risks to clinical safety and efficacy should be addressed based on the nature and degree of those differences.” According to Ritter, bioprocessors could benefit from continued advances in sensitivity and specificity of physical and functional characterization tools, particularly those used to assess posttranslational modifications and higher-order structure, as well as potency (through improved in vitro bioassays). She would also like to see more user-friendly analytical tools for generating accurate, reliable, robust results. “Many analytical instrument and assay development companies focus only on R&D applications,” she explains, “but they fail to recognize the tremendous value added when their improvements are also designed to meet the operational needs of a regulated QC-testing laboratory. The analytical data from such labs allows each and every batch of a biotech product —originator or biosimilar — to be used in patients. So the performance of the analytical techniques and instruments in a QC environment is vital.” |
Art or Science?
Developability assessment for all therapeutic proteins considers their physicochemical properties, with complex quality profiles and by addressing formulation issues such as aggregation and viscosity. The issues for new molecular classes such as bispecific antibodies or antibody–drug conjugates (ADCs) are less well understood and probably less easily transferred from one process to another than more familiar biologics (e.g., MAbs).
“Although early stage molecule optimization focuses on the underlying DNA sequence and molecular properties, both expression host and process conditions affect the protein produced and decisions taken,” notes Barney Zoro, product manager at TAP Biosystems (Royston, UK), a Sartorius Stedim Biotech company. During cell-line screening, the goal is optimizing gene delivery with minimal impact on host-cell performance. Also, good control of process conditions is critical to accurate scalability and prediction of manufacturing performance — and hence to accurate selection decisions.
Those critical goals can be achieved through microbioreactors with pH and DO control, impeller agitation, gas sparging, and automated liquid additions at times that are suitable for the process. The similarity of, for example, TAP’s ambr minibioreactor systems to production bioreactors delivers a better match of protein production outcomes compared with shaken-type systems. Microbioreactors with high-throughput capacity permit study of a much wider range of conditions that closely approximate those at manufacturing scale. Zoro notes that eighteen of the top 20 biopharmaceutical companies use ambr products for early stage expression, CHO clone screening, and media and process optimization studies.
Whereas smaller-volume microbioreactors in the 15-mL volume range support early stage clone selection, process development is more reliable at larger volumes (e.g., the ambr250 system’s 250-mL volume). “Larger culture volumes support upstream bioprocessing and generate enough material for downstream development studies,” Zoro says, “thereby allowing investigation of process conditions on overall developability and whole- process performance.” Larger systems also allow full DoE capabilities, with data acquisition and management for such complex studies. “This approach provides a solid foundation for operating a process within the desired developability range for both optimum outcomes and process robustness.”
Scale-up remains more art than science, says Govind Rao, director of the Center for Advanced Sensor Technology at the University of Maryland, Baltimore County. This is in part because of difficulties in integrating existing sensor technology with single-use systems, but also because most sensing and monitoring methods largely ignore events at the cellular level.
Rao and his colleagues have used quantitative polymerase chain reaction (qPCR) for metabolic monitoring. (Editor’s Note: See the article by Baghbaderani, Rao, and Fellner in this month’s cell-therapy supplement.) In collaboration with the FDA scientists, they have identified “sentinel” genes associated with critical cellular activities such as apoptosis. Amplifying those genes indicates which of the sentinels are up- or down-regulated, thus providing a measure of culture health. As determined by qPCR, gene expression and therefore cellular status are independent of scale. So gene expression can indicate whether process conditions are preserved during scale-up or scale-down. The technique, Rao claims, also applies to clone selection and cell-line development. The downside is that it requires sampling, which bioprocessors typically resist for reasons of potential contamination. But Rao believes that sampling would be “well worth it” as a precondition of obtaining “enormous insight” regarding cellular metabolism.
“These sentinel genes allow you to zoom in on the cellular metabolic state,” he says. Conventionally, process designers measure gross parameters such as oxygen transfer rate in bioreactors at scale-up/scale-down. “But bubble distribution is different, and depending on the size of the tank, the head pressure is different. So to accurately represent a 10,000-L tank, the scaled-down system needs a very smartly designed minibioreactor that reflects what’s going on at manufacturing scale.” Using sentinel genes, process engineers can design the physical hardware to ensure a consistent cellular metabolic response.
| Scale-Down Systems for Early Assessment of Manufacturing Production process engineers use automated microplate- and liquid-handling systems to screen for clonal productivity and high-level process parameters related to media and feed. Bench-scale bioreactors in the 2-L to 5-L volume range more closely resemble ultimate process conditions, but they take up too much space and consume too many resources to be truly parallel and high-throughput.Between those options are small, parallel bioreactor systems that mimic large-scale conditions to varying degrees.Biomanufacturers are adopting these systems for screening conditions, media, feed options in process development; for process-parameter optimization, microproduction of relevant proteins in research quantities, and for “scale-down” trouble-shooting and validation experiments.The TAP Biosystems (a Sartorius Stedim Biotech company) ambr15 and ambr250 microbioreactors provide 15-mL and 250 mL working capacity, respectively. They are arguably the best known micro- and minibioreactors, but they are not the only option.Germany’s m2p-labs offers two models. Its BioLector microbioreactor platform for high-speed bioprocess development features 48 parallel microplate bioreactors, with online condition monitoring and process control in a disposable platform. It uses optical sensors, and m2p-labs reports on applications for both aerobic and anaerobic cultures. The company’s Robolector CM robotic microbioreactor offers more comprehensive monitoring of cell culture parameters, making it suitable for quality by design (QbD) and design of experiment (DoE) use.Applikon Biotechnologies in The Netherlands taglines its MiniBioreactor systems as “real small bioreactors.” They hold process volumes ranging from 250 mL to 1 L — defining the approximate midpoint between typical minibioreactors and bench-top laboratory systems. The larger working volumes and benchtop footprint come with exquisite control over process parameters. According to Applikon, MiniBioreactor units may be “customized to fit the demands of any process.”Pall Life Sciences offers its Micro-24 MicroReactor system along similar lines. This runs aerobic and anaerobic microbial fermentation as well as mammalian and insect cell cultures while monitoring and controlling each of 24 reactors’ gas supply, temperature, and pH. Sensors for pH and DO are precalibrated for 3-mL to 7-mL working volume microreactors.Those systems all mimic batch and fed-batch processes. Pharyx Inc. of Woburn, MA, has developed perhaps the smallest-volume microbioreactors of all (1 mL). These are suitable for batch, chemostat, and turbidostat cultures, with or without perfusion. Combining perfusion with chemostat or turbidostat modes allows control over secreted product concentrations independent of cell dilution. The reactors are prefitted with sensors for pH, DO, and optical density. Thanks to microfluidic flow designs, they provide five fluid feeds or four feeds as well as perfusion options. |
From DoE to QbD
Design of experiments and quality by design have been instrumental in supporting developability and manufacturability assessments. DoE includes statistical tools aimed to help scientists understand complex problems that are defined by multiple and sometimes interdependent variables and thus not amenable to traditional “one-variable-at-a-time” experimental approaches. Because of the connection between DoE and aspects of process understanding, some people identify it with QbD. But the latter is far more ambitious.
“QbD looks foremost at the design criteria for a product to fulfill a number of performance requirements — therapeutic, economic, logistic, clinical, and so on — that will define a target product profile (TPP),” Zurdo explains. “From that, scientists can derive specific critical quality attributes (CQAs) with associated risk assessments and risk mitigation plans to follow.”
Process understanding begins after initial product design, he says, by mapping how a process influences specific product quality attributes. “Unfortunately, most attempts at implementing QbD in biopharmaceuticals have been reduced primarily to process understanding and its influence in a handful of CQAs, largely linked to chemical identity and stability. However, the real scope of QbD is much broader. Its implementation requires involvement of many stakeholders with different responsibilities in the therapeutic development continuum, from laboratory to clinic.”
Four Key Components: QbD incorporates a proactive approach that requires identification of all critical material attributes and process parameters, then determining the extent to which variations could affect product quality. The more information generated on the impact — or lack thereof — of a component or process on a product’s quality, safety, or efficacy, the more flexibility can be allowed during manufacturing.
Establishing a design space does not imply testing ad infinitum, says Aranha. To keep from exploring every permutation and combination of materials and processes, developers apply tools such as multivariate analysis. “This allows evaluation of process perturbations during actual manufacturing operations, where they are likely to occur. That helps identify the root cause and proceed with deviation resolution, depending on whether or not it falls outside the design space.”
Aranha says QbD has four key components: homing in and defining a product-design goal, establishing a process design space, setting control-space parameters, and defining or setting operating-space limits. Additionally, the practice is based on a few intuitively obvious prerequisites for success: process knowledge, financial investment, risk and project management, and regulatory input.
Foremost is process knowledge and competence. DoE studies, for example, provide myriad data that should be analyzed properly by experts (statisticians) because decisions based on them significantly affect development.
Next comes financial investment. Return on investment (RoI) may not be immediately obvious for QbD, but its true potential is ultimately to improve the bottom line. “This aspect is often a significant hurdle,” Aranha says. “Many companies are not willing to invest based on future returns.”
Leveraging available risk-management and project-management tools is the third precondition. Although such tools and statistical packages abound, developers must choose wisely based on specific requirements of their molecules in development.
The final prerequisite is open communication with regulators, which includes sharing of process knowledge. Risk assessment should incorporate not just traditional scientific and regulatory factors but business feasibility and return on investment.
That being said, Aranha cautions that QbD does not replace the need for process validation and GMP compliance. “Rather, it is a complementary methodology based on sound scientific principles that should leverage product and process knowledge to streamline drug development.” Increased process understanding through application of statistical tools such as multivariate analysis and DoE studies (together with robust risk management) can steer a control strategy during process validation.
QbD is still not mandated by regulation, but adopting it can bring multiple benefits, even after product approval. Aranha summarizes the scenario as “pay me now, or pay me later.” If the design space is well understood, then evaluating and remediating out-of-specification (OoS) results become more straightforward activities. Moreover, the business benefits are tangible: fewer lost batches, fewer manufacturing deviations, faster time to market, more reliable clinical and commercial supplies, and fewer regulatory hurdles, which translate into a many-fold RoI through direct cost savings and increased revenue.
Aranha advises developers to take advantage of today’s advanced bioinformatics and computational power to ask complex genomic and proteomic questions. “An essential component of QbD is determining the desired target product profile (TPP) rather than relying on traditional hit-and-miss or ‘shotgun’ approaches,” she explains. “Chromosome walking and other advanced methodologies allow us to query and zero in on desired characteristics and focus on target genes for therapeutic applications.”
Focusing on Manufacturability
Whereas small-molecule drug manufacturability relates mainly to chemical-bond formation and purification, a significantly greater number of inputs contribute to or detract from biopharmaceutical manufacturability. Although developability and manufacturability overlap, they are often considered distinct. The degree of overlap is telling, however.
At Novartis, full developability assessment occurs through an integrated R&D approach. The IBP team uses expression technology and manufacturing processes that are as close as possible to those implemented for final production. Manufacturability assessment testing therefore is not a separate effort.
Novartis is not alone in that philosophy. Zurdo at Lonza also believes in connecting developability and manufacturing process development. “The main motivations should be to speed development and reduce costly failures,” he emphasizes. “Practically, this means first integrating discovery (lead selection and optimization activities) with process development and implementing methodologies that facilitate risk assessment with minimal material requirements and maximal throughput.”
Ideally, combining computational models with surrogate analytics (methods that inform about quality attributes without requiring detailed studies) provides a platform for assessing multiple options inexpensively within a compressed timeframe. “Moving one step forward,” Zurdo says, “we could imagine how, by mapping the behavior of a lead candidate, we might also enable more effective process design.” For example, aspects of product stability are usually not addressed until a bioprocess has been defined. By introducing such considerations earlier in development, companies might prevent conditions that could negatively affect stability during manufacturing.
Late Fine-Tuning: BPI editorial advisor Michiel Ultee of Ulteemit BioConsulting agrees that companies will find ways to overcome aggregation or low titer. “You can always go to larger tanks or remove aggregates during downstream processing.”
One of his clients was forced to change the primary sequence in the constant region of a MAb, where allotropic variants are common, as opposed to the all-important binding region. This particular molecule contained a proline residue. Because proline is cyclic, it produced a kink in the protein’s primary structure that was responsible for low titers and aggregation. When a linear amino acid was substituted for that proline, productivity jumped sixfold with significantly reduced aggregation.
Ultee also has seen difficult-to-manufacture fusion proteins for which developers plowed ahead into phase 1 and worried about cell line and process changes later. “The pressure to get into the clinic is so high,” he explained, “that even if it’s not the most robust process, you’ll use it for that purpose.”
Manufacturability assessments based on yield are highly dosage-dependent. Another Ulteemit client was developing a fusion protein that exhibited poor yield and high aggregation. Yet the sponsor forged ahead into phase 1 at the 2,000-L scale. “Luckily, the dosage was below one milligram,” Ultee reports, so the company was able to get by with what is normally considered a low-yield process. But at phase 2, it had to reengineer its cell line for higher productivity.”
The fine-tune–late strategy is becoming increasingly popular. It requires companies to keep process and clinical development in synch, with the latter particularly responsive. With processes more or less set in stone by the initiation of phase 3 studies, the timing for this smart strategy must be near perfect.
“If you optimize a cell line and molecule too early and a product fails in phase 1, then you’ve spent a lot of resources on a failure,” Ultee explains. “It’s a balance: Would you rather expend resources developing a robust process or use an adequate process that will probably not stand the test of time, but which will at least get you into the clinic? Successes in human subjects are what generate investment. Animal studies are required, but they don’t convince a lot of investors to open their wallets.”
Note that although a phase 3 process may differ from that used to produce first-in-human (phase 1) material, the product must be at least as pure and as active as that made by earlier processes. “If you’re down from 200 ppm to 75 ppm host-cell proteins,” Ultee adds, “then there will be no arguments.”
| Less Becomes More: Benefits of High Titers and Low Dose With the great clinical and commerical success of monoclonal antibodies (MAbs) throughout recent decades, parameters are well established for assessing manufacturability of such immunoglobulin proteins. MAbs are generally high-dose products manufactured by high-titer expression-host systems with predictable purification schemes. Because platform processes are rare for nonantibody biologicals (1), evaluating their manufacturability involves such factors as dosage size, market size, volumetric productivity, and final drug cost — how much you need, how much you can produce, and how much you can charge for it.Rising production titers have been very good for MAb manufacturability considerations. “Instead of a 20,000-L tank,” comments Michiel Ultee of Ulteemit BioConsulting, “you can get by on a 2,000-L bioreactor, even less if you have a niche product. But you still need a sound, robust process.”Emerging biopharmaceuticals such as antibody–drug conjugates (ADCs), fusion proteins, and enzymes probably will be significantly more potent than conventional MAbs. That potentially lowers the bar for their manufacturability in terms of drug-product requirements. However, lower expected volumetric productivities for nonplatform molecules may zero out the otherwise benefits of lower dose.Market size plays significantly in the manufacturability equation. At the low end of this spectrum are niche products, orphan drugs, and personalized medicines. “In these situations,” Ultee explains, “you don’t need as much material. But these drugs are not often ‘treat-and-done’ products; they’re usually administered regularly for life, which raises material requirements.”Pricing affects manufacturability directly. With dosing quantity and frequency, dosage size, and market size being equal, developers can be more forgiving of process or production shortcomings for a $100,000 therapy than for a $10,000 treatment. But companies don’t always get their calculations right. “Dendreon’s Provenge immunotherapy for advanced prostate cancer is expensive and only extends life a few months,” Ultee explains. “Hence, the marketplace has not been enthusiastic. On the other hand, you have Gilead’s hepatitis C drug (Harvoni), which is also expensive, but is curative.” In the latter situation, the market valued the drug based on its efficacy and lower total cost relative to alternatives, which are chronic disease and repeated hospitalizations. Reference 1 Scott C. Toward Nonantibody Platforms. BioProcess Int. 10(10) 2012: 31–42. |
A “Wholistic” Approach: When encountering difficulties in a particular process step, developers should examine the process as a whole rather than focusing on the step at hand, says Claire Scanlan, process development scientist at EMD Millipore (Billerica, MA). “A problem may be resolvable further upstream in the process.”
For example, increasing titers in MAb processes will generate higher impurity levels (especially DNA and host-cell proteins), which can challenge standard purification templates and platform processes or tax the purification infrastructure of existing hardware. Acid precipitation or addition of cationic flocculation polymers at harvest can reduce impurities, lessen the load on an existing purification train, and perhaps increase resin lifetime.
Similarly, increased aggregate concentrations in higher-titer MAb process streams also can lower capacities in virus filtration, an extremely costly biopharmaceutical unit operation. “Further optimization of upstream purification and/or the implementation of an adequate prefilter can significantly improve performance of the virus filter,” Scanlan says. She advises bioprocessors facing such challenges to consult with filter or resin vendors for help finding solutions. “These efforts can be mutually beneficial.”
Several EMD Millipore products, including newer media grades for the company’s Millistak+ HC depth filters, have resulted from collaborations with companies that have encountered issues that the company’s product portfolio at the time failed to address. “Examine all aspects of the process and potential partnerships when dealing with troublesome unit operations,” Scanlan says. “Keeping a wider view can yield exponential benefits at the step of interest.”
Good Enough Is Good Enough: With bispecific antibodies, developability (and later manufacturability) depends on early molecular design. “Without it, the combination of heavy and light chains would result in 10 isoforms that would be inseparable using conventional downstream processing,” notes Guenter Jagschies, senior director of GE Healthcare Life Sciences. “It would be a mess that would not be developable.”
Jagschies points to the emergence of techniques that direct specific pairings of heavy and light chains: for example, Roche’s CrossMAb technology, which prevents formation of unwanted side products. That works specifically through the light chain. So-called “knob and hole” technology developed at Genentech in 1997 directs desired pairings of heavy-chain elements. “Without techniques like these,” Jagschies says, “bispecific antibody products would not exist.”
Hand-in-hand with molecular design goes selection of an expression system that allows developability assessment of multiple candidate designs in parallel using manufacturing models that are predictive of a final process. In 2014, GE Healthcare in-licensed intellectual property specific to Chinese hamster ovary (CHO) cells from Promosome. The technologies include translational enhancer elements for difficult-to-express proteins, landing-pad technology for rapid cell line development, and signal peptides optimized by synthetic biology to improve processing of a broad range of proteins. GE plans to offer these technologies as part of its cell line development services. “Together they will enable us to assess developability in a more predictive, high-throughput, and parallel fashion,” says GE’s Daniel Ivansson.
Similarly, Regeneron has developed a suite of tools for controlling productivity and quality. Its EESYR service provides site-specific integration into a transcriptional “hot spot” for rapid isolation of highly expressing cells; the FASTR service selects cells that secrete the highest levels of recombinant proteins; and the NICE service delivers control of secreted-protein expression. The company claims that the EESYR approach reduces cell line development time from several months to several weeks. That service is already used in current GMP (CGMP manufacturing.
“Companies can obtain titers of up to about 3 g/L, which is sufficient for up to phase 2 studies,” Jagschies observes, “without having to return to molecular design to improve developability or manufacturability.” He calls this the “good-enough” approach to product development, a strategy that’s on the rise at top companies to delay investment in full production-scale processes until favorable study results are in hand.
Supply As a Manufacturability Input: Availability of supplies and ingredients is an oft-overlooked component of manufacturability and developability. For single-use bioprocessing, major concerns include reliable access to consumables (e.g., bags, tubing, and connectors) and a general lack of interoperability among different suppliers’ offerings.
| Working with Downstream Customers
Several EMD Millipore products have evolved from specific customers’ identified needs and/or process technology gaps. For example, Express SHF (sterile high-flux) hydrophilic cartridge filters were developed after one customer identified a need for a more economic, faster-flowing, and caustic-compatible sterilizing-grade filter for chromatography buffers. And F0HC-grade Millistak+ disposable depth filters were developed with a customer to clarify a specific acid-precipitated monoclonal antibody (MAb). Because acid addition shifted the particle-size distribution of that process fluid, the company’s other depth-filter offerings weren’t optimized to capture the altered product stream. “These relationships were mutually beneficial,” notes Clare Scanlon. “The customers helped identify a product technology gap, which was probably also prevalent in other customers’ processes. And the customer allowed EMD Millipore to conduct alpha and beta test samples using its own process feed to ensure that the product would fully fit the customers’ (and industry’s) growing needs.” Customers also should examine the manufacturing process as a whole. Instead of increasing the size of a protein A column to accommodate higher impurity levels in a high-titer MAb process, Scanlon advises customers to look upstream at reducing impurity levels present before MAb feed is loaded onto the capture column. Possibilities for intervention include acid precipitation or polymer flocculation or addition of a depth filter on the protein A load. “That is often more cost effective than using more protein A resin,” Scanlon says “and can also increase the lifetime of the protein A column.” |
A similar situation exists for cell culture media, which often include hundreds of ingredients and components. Virtually all processes for MAbs and nonantibody therapeutic proteins alike now use media that are free of animal components. Many companies are switching to products based on plant protein hydrosylates (peptones), whereas some instead use chemically defined (CD) media, in which all components and their concentrations are fully quantified.
Because the ingredients of animal-component–free and CD media may number in the hundreds, such products represent an ideal test case for supply chain reliability. Leading vendors of cell culture media routinely verify their ingredient suppliers several levels down to assure raw material quality and availability for their customers.
Here is where change management becomes critical. Thermo Fisher, for example, maintains a list of approved ingredient suppliers who are required to provide change notifications regarding source materials or manufacturing processes. Thermo Fisher assesses the impact of those changes and notifies its own customers when appropriate. The company is also open to quality and technical visits by those customers. A quality audit could include the ability to inspect Thermo’s own manufacturing facility and obtain access to batch records for products covered by the supply agreement.
| Antibody Engineering for Stability Enhancement by Rajiv PanwarAntibodies are the leading class of drugs entering clinical trials. Despite their success, many challenges come with antibody manufacturing. Considerable attention has been paid to improving the stability of such products through buffer and excipient optimization. Doing so, however, is time consuming and arduous. Alternative approaches are needed to reduce the time and effort needed to make an antibody stable under different stress conditions.One option that is beginning to show promise is antibody engineering. This approach has potential to improve antibody stability at the discovery stage, thereby saving a significant amount of development time. The constant domains are known to have limited diversity, whereas much sequence diversity is found in variable domains. Although variable domains are known to have more aggregation propensity, a number of engineering approaches have been attempted to make both domains stable.Preliminary work by Demarest, et al. to predict stabilizing mutations used sequence analysis where they identified 371K, 376G, and 392L as being responsible for thermal instability (1). The team significantly improved thermodynamic stability (76–86 °C) of the CH3 domain through residue substitution.In recent years, camelid and shark antibodies have attracted much interest with their exceptional thermal stability. It has been attributed to the presence of an additional disulfide bridge and long CDR loops (2). On similar lines, disulfide intradomain bridges were inserted in the CH3 domain, which increased the thermal stability of the Fc fragment (3, 4).Stability improvements based on charge introduction can be an effective approach. A significant improvement in aggregation resistance (more than 35-fold for both VH and VL) was observed when aspartate was introduced in the CDR regions (5). A potent IgG1 monoclonal antibody suffered from poor expression, heterogeneity, and aggregation, making it difficult to scale up for clinical trials. Structural modeling revealed that a highly exposed cysteine residue was responsible for that antibody’s instability. By replacing cysteine with threonine, Buchanan, et al., significantly reduced the antibody’s aggregation, improved its homogeneity, elevated the product’s melting point (Tm), and enhanced expression levels by 26-fold (6).Other researchers have tried to find mutations in the VH or VL antibody domains that are responsible for increasing the folding stability. In one example, Barthelemy, et. al. randomized 20 residues at the VH–VL interface on a VH domain to identify stability enhancing mutations (7). They selected four mutations that substantially increased the folding stability of the VH domain. Those mutations also resulted in favorable orientation of residues responsible for better folding stability. Additional stabilization approaches include humanization or grafting of complementarity-defining regions (CDRs), hydrophobic-residue swaps, addition of sugar moieties, enhanced interactions between VH and VL domains, and introduction of constant domains. All have been tried to increase the kinetic stability of antibodies (8). Many engineering approaches have proven to be useful for increasing overall antibody stability. However, when engineering an antibody to improve its biophysical characteristics, it is also essential to preserve the molecule’s binding affinity without introducing unwanted variations that could lead to immunogenicity. References 1 Demarest SJ, et al. Optimization of the Antibody CH3 Domain By Residue Frequency Analysis of IgG Sequences. J. Mol. Biol. 335, 2004: 41–48. 2 Ewert S, et al. Biophysical Properties of Camelid V(HH) Domains Compared to Those of Human V(H)3 Domains. Biochem. 41, 2002: 3628– 3636. 3 Wozniak-Knopp G, et al. Stabilization of the Fc Fragment of Human IgG1 By Engineered Intradomain Disulfide Bonds. PLoS One 7, 2012: e30083. 4 Ying T, et al. Engineered Soluble Monomeric IgG1 CH3 Domain: Generation, Mechanisms of Function, and Implications for Design of Biological Therapeutics. J. Biol. Chem. 288, 2013: 25154–25164. 5 Dudgeon K, et al. General Strategy for the Generation of Human Antibody Variable Domains with Increased Aggregation Resistance. Proc. Natl. Acad. Sci. USA 109, 2012: 10879–10884. 6 Buchanan A et al. Engineering a Therapeutic IgG Molecule to Address Cysteinylation, Aggregation, and Enhance Thermal Stability and Expression. MAbs 5, 2012: 255–262. 7 Barthelemy PA, et al. Comprehensive Analysis of the Factors Contributing to the Stability and Solubility of Autonomous Human VH Domains. J. Biol. Chem. 283, 2008: 3639–3954. 8 Perchiacca JM, et al. Engineering Aggregation Resistant Antibodies. Annu. Rev. Chemi. Biomol. 3, 2012: 263–286. Rajiv Panwar is a scientist II at Akrivis Technologies, LLC; One Broadway 14th Floor Cambridge, MA 02142; 1-617-401- 3390, fax 1-617-401-3613; rajpanwars@gmail.com; www.akrivis.com. |
Total Failure Is Rare: According to Wheelwright, it is rare for products to fail due to manufacturing difficulties. “The question is not whether a drug can be manufactured,” he explains, “but rather how long development will take and what recovery is possible during manufacturing. These factors affect costs of development and of manufacturing. The challenge is when to make a decision to stop development and accept the results one has achieved.”
After all, the principal endpoints for licensing are safety and efficacy. “Even when your process is variable,” says Serena Smith (process science manager at Thermo Fisher Scientific), “if you can prove that it is in control and that the product is safe and effective, regulators will generally be satisfied.” Beyond that, go–no-go decisions come down to business factors, such as cost of goods and market considerations.
“In my past role developing biological processes at a large pharmaceutical company,” Smith continues, “I observed projects being canceled at various stages of development. Some projects were terminated before animal toxicity studies (likely because of a lack of market demand). Other projects ended at various clinical phases based on safety or efficacy data.”
Many factors and inputs play into go–no-go decisions, including anticipated time to market, competition from innovator molecules and generics, and market size. Developers may decide to lower a drug’s pipeline priority or sell it to a competitor. Regardless, as developability and manufacturability coalesce, the latter may lose some relevance as a distinct consideration for biopharmaceuticals.
For Further Reading
Agrawal NJ, et al. Aggregation in Protein-Based Biotherapeutics: Computational Studies and Tools to Identify Aggregation-Prone Regions. J. Pharmaceut. Sci. 100(12) 2011: 5081–5095.
Baker MP, et al. Immunogenicity of Protein Therapeutics: The Key Causes, Consequences, and Challenges. Self/Nonself 1(4) 2010: 314–322.
CBER/CDER. Guidance for Industry: Immunogenicity Assessment for Therapeutic Protein Products. US Food and Drug Administration: Rockville, MD, August 2014.
Jarasch A, et al. Developability Assessment During the Selection of Novel Therapeutic Antibodies. J. Pharmaceut. Sci. 104(6) 2015: 1885–1898.
Kondragunta B, et al. Bioreactor Environment-Sensitive Sentinel Genes As Novel Metrics for Cell Culture Scale- Down Comparability. Biotechnol. Progr. 28(5) 2012: 1138– 1151.
Kondragunta B, et al. Genomic Analysis of a Hybridoma Batch Cell Culture Metabolic Status in a Standard Laboratory 5-L Bioreactor. Biotechnol. Progr. 28(5) 2012:1126–1137.
Nichols P, et al. Rational Design of Viscosity Reducing Mutants of a Monoclonal Antibody: Hydrophobic Versus Electrostatic Inter-Molecular Interactions. MAbs 7(1) 2015.
Tai M, et al. Efficient High-Throughput Biological Process Characterization: Definitive Screening Design with the ambr 250 Bioreactor System. Biotechnol. Prog. 3 July 2015; e-pub ahead of print.
Van De Weert M, Møller EM. Immunogenicity of Biopharmaceuticals. Springer Science and Business Media: New York, NY, 2 June 2008.
Zurdo J, et al. Early Implementation of QbD in Biopharmaceutical Development: A Practical Example. BioMed Res. Int. 2015; www.hindawi.com/journals/ bmri/2015/605427/cta.
Zurdo J. Developability Assessment As an Early De-Risking Tool for Biopharmaceutical Development. Pharmaceut. Bioproc. 1(1) 2013: 29–50.
Zurdo J, et al. Improving the Developability of Biopharmaceuticals. Inno. Pharmaceut. Technol. 37, 2011: 34–40. c
With a PhD in organic chemistry from the State University of New York at Stony Brook, freelance writer Angelo DePalma (angelodp@gmail.com) was a chemist first at Brookhaven National Laboratory and then at Schering- Plough. For over two decades, he has written for dozens of technical online and print publications, as well as product and service companies in biotechnology, bioprocessing, pharmaceutical chemistry, pharmaceutical development, drug discovery, and laboratory instrumentation.