Quality by design (QbD) is a systematic approach to drug development. It begins with predefined objectives and emphasizes product and process understanding and process control, all based on sound science, data-based decision making, and quality risk management (QRM). As introduced by the US Food and Drug Administration (FDA), QbD brings modern drug development methodologies to chemistry, manufacturing, and control (CMC) teams working on biologics, pharmaceuticals, and vaccines. The innovations associated with QbD are not so much the development concepts (which have been proven in multiple industries for well over 20 years). Rather, such innovations are the application of those principles in the development, submission, and manufacturing of drug products and drug substances.
QbD was introduced over 10 years ago; however, regulatory agencies are now in the process of moving it from recommended (optional) to mandatory in drug submissions and filings. QbD has the following 10 guiding principles:
-
A clear line of sight from clinical to product release and stability
-
QRM in every aspect of development
-
Enhanced product understanding
-
Assay understanding
-
Process understanding and characterization
-
Generation of transfer functions
-
Improved product specification limits and justification
-
Robust design space and edge of failure
-
Use of modern control strategies and process analytical technologies (PAT) (1)
-
Continuous improvement and validation throughout a product’s lifecycle.
As stated in the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) Q8 Pharmaceutical Development guideline, Section 2 (2):
The aim of pharmaceutical development is to design a quality product and its manufacturing process to consistently deliver the intended performance of the product. The information and knowledge gained from pharmaceutical development studies and manufacturing experience provide scientific understanding to support the establishment of the design space, specifications, and manufacturing controls.

There must be a clear definition of all product requirements and performance characteristics of a drug. Information for CMC teams and others regarding clinical indication, stability, product form, dose, method of administration, and other product requirements are stated in a quality target product profile (QTPP). Critical quality attributes (CQAs) are defined early in a development process, and they should be managed and updated throughout a product’s life cycle. A complete list of analytical methods associated with CQAs help to clarify the line of site from CQA to assay (Figure 1).

Systematic drug development and QRM ensure that a line of site is maintained from a target product profile to the QTPP. It continues through CQA generation and analytical method selection, unit operation definition and characterization, specification and acceptance criteria limit generation, critical process parameter (CPP) and control selection, then finally method and process validation. ICH Q6, Q8, Q9, Q10, and Q11 documents (2,3,4,5,6) detail the pharmaceutical development process and how to set limits. The QRM process is linked to product and process CQAs because they are the drug quality attributes we wish to maintain.
QRM in Every Aspect of Product DevelopmentQRM is an essential element of drug development and manufacturing throughout a product’s life cycle. QRM as stated in ICH Q9 is designed to ensure that drug CQAs are defined and maintained from phase to phase during drug development and manufacturing. It’s meant to secure that changes in drug product formulation, definition, analytical methods, and associated process changes are understood and managed to ensure patient safety and drug efficacy. An effective QRM process can help manufacturers produce high-quality drug products by providing a proactive means to identify and control potential risks to quality during development and manufacturing. Customer and market understanding, product definition and understanding, and process understanding are all the main objectives of QRM throughout all development activities (Figure 2).

Two key QRM principles are as follows:
-
Risk assessment should be based on scientific knowledge associated with product and process understanding
-
The level of effort and detail associated with risk assessment and management should be commensurate with the level of risk being identified and evaluated.
Science guides us, and facts and data minimize and help control risk. The major benefits of QRM and risk assessments are to improve manufacturers’ ability to develop drug products and drug substances and to answer the questions of when, what, how much, and where additional development is needed. Development activities reduce potential risks to safety and efficacy and help manufacturers achieve the benefits of a drug for a targeted indication.
Enhanced Product UnderstandingThe primary drug manufacturer must understand all critical and key multiple factors influencing your product and all primary sources of variation. The three parts of product understanding are system design, parameter design, and tolerance design.
System design is the selection of a technology, chemistry, active pharmaceutical ingredient (API), and materials as well as an understanding of impurities found in a drug product an
d drug substance.
Parameter design is the selection of key targets, set points, and concentrations.
Tolerance design is the allowable range or limits for each parameter. Capability assessment indicates how well you can meet defined tolerances. The enhanced approach is systematic, with a mechanistic understanding of the multivariate factors influencing products.
Assay UnderstandingCare must be taken to properly select analytical methods for measuring all CQAs. Each selected assay must be fit for use and valid for the condition to be measured. During method development, key factors and process steps that influence method variation must be identified, optimized, and controlled. Experimental design and partition of variation can be used to understand and minimize assay variation.
Precision, accuracy, linear range, limit of quantitation (LOQ), limit of detection (LOD), and percent of tolerance must be defined and validated during method development. Reference standards and assay controls must be determined to ensure that data generated from the assay are dependable and do not confuse product and process variation with variation from the analytical method. Relative standard deviation or coefficients of variation alone are not sufficient measures of assay accuracy. They should be used with other measures of assay validity and systems suitability. Accuracy to precision profilers should be used to explore assay design space and visualize whether a method is acceptable relative to product limits (Figure 3).
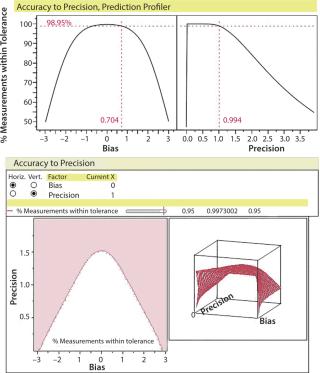

Factors influencing a production process and associated variations should be understood. Process understanding has the same three components as product understanding: system, parameter, and tolerance design. Process understanding is a prerequisite to stage 1 process validation.
With a QbD approach, some process parameters may be adjustable within a defined design space. Many processes have both dynamic and static properties. Understanding and controlling process dynamics is at the heart of an enhanced approach and will reduce variation at the product and process target. CPPs and their associated effect size must be identified and controlled during process development.
Generation of Transfer FunctionsManufacturers should understand how X factors influence Y responses, where each is in the form of an equation and either reflect scientific knowledge or be empirically derived from structured multiple factor experimentation. Ultimately, knowledge should be expressed in the form of an equation to be fully useful (
Equation: Transfer function from a drying process

Many software packages such as SAS/JMP will generate the transfer function during process characterization and experimentation.
Improved Product Specification Limits and JustificationSpecification limits must be a part of an overall product control strategy. They should be linked to CQAs and the QTPP (line of sight). These limits should be based on scientific knowledge, transfer functions, and/or statistical distributions. Ideally, a transfer-function approach to setting limits is best. Using statistical distributions to set limits is a reasonable approach when all unit samples indicate good safety and efficacy and no harm relative to clinical outcomes. If edges of failure are detected, then statistical distributions should not be used.
Robust Design Space and Edge of FailureICH Q8 defines design space as “The multidimensional combination and interaction of input variables (e.g., material attributes) and process parameters that have been demonstrated to provide assurance of quality” (2). That definition, however, has a logical flaw: Design space is a visualization of the average performance at fixed set points. In truth, it often may misrepresent the edges of failure and the actual process dynamics at play during processing. Simulation and edge-of-failure analysis augment design-space visualization to ensure that the allowable range of variation will cause no harm and will not result in out-of-specification (OOS) conditions.
Design space is proposed by an applicant and approved by a regulatory agency. A design space allows a manufacturing operation to adjust parameters within the space to improve drug substance consistency and control. Proven acceptable ranges and normal operating ranges must be set and visualized relative to the particular design space.
Control Strategies and PATControls are defined to ensure potency, purity, safety, and efficacy. The three parts of a control strategy are in-process test and release, postprocess test and release, and closed-loop process measurement and adjustment during processing. The first two are traditional, but closed-loop in-process controls are relatively new to good manufacturing practice (GMP) operations. Adjustment of temperature, pH, time, and amount of a key ingredient are all examples of enhanced closed-loop controls.
Closed-loop controls need four parts: sensors to detect deviation from target in a product or process, alarms to determine when to adjust and when to leave alone, control logic and adjustment rules (how much adjustment to make), and verification that adjustments are correct and effective. The PAT framework is a good guide for how closed-loop controls should be defined and validated.
Continuous Improvement and ValidationTracking and trending have generally been the method for monitoring drug products and drug substances to assess lot variation and process trends. Properly designed, such systems and controls will lead to preventative action rather than corrective action before OOS events and lot failures.
Measures of process capability, variation, and controllability will continuously validate a process and demonstrate it is fit for use. They also provide acceptable process outcomes.
Closing the GapsQbD is a modern, updated, and enhanced method of drug development, manufacturing, and control. It closes many gaps associated wit
h traditional drug development and provides higher levels of quality assurance, reduced variation, and improved control.
The industry is currently experiencing a “knowledge and experience deficit” regarding the use of QbD principles for many CMC teams. Until manufacturers have successfully used QbD, achieved positive regulatory submissions, and produced good outcomes in their GMP operations, they will have some degree of fear or uncertainty in QbD’s use. Consulting and training are typically used to fill the gap until the teams have sufficient experience to do the development on their own.
The QbD framework for drug development became mandatory in 2013 from the FDA and EU. For now, completing a design space is optional.
Author Details
Thomas A. Little, PhD, is president of Thomas A. Little Consulting, 12401 North Wildflower Lane, Highland, UT 84003; 1-925-285-1847;