Commercializing cell therapies can be much more challenging than commercializing traditional pharmaceuticals and biologics. Cell-based drug products are significantly more complex than protein or small-molecule drug products. Their mechanisms of action and product attributes are also more complex. Cell therapy product attributes rely heavily on the associated manufacturing processes. Process changes can influence products in ways that may not be discernible until their effects on efficacy effects become evident. Product characterization is critical, but cell therapy products are living organisms with many characteristics. For pharmaceuticals and biologics, planning and process changes for commercial manufacturing often can wait until clinical results are available. But for cell therapies, changes during planning and processing carry a much higher risk. Companies may be unable to determine the effects of such changes on product efficacy without needing more clinical data.
As a result, planning a cell therapy’s commercial manufacturing strategy must begin much earlier than such planning for pharmaceuticals or biologics. Doing so prevents substantial delays, additional costs, and even commercial failure after clinical success.
Only a handful of cell therapies have been commercially approved to date, reflecting both the complexity of this therapeutic approach and the relative youth of the industry. The number of cell therapies currently in phase 2 and phase 3 development and delivering promising results, however, evidences the industry’s advancement toward a commercial state. However, US FDA approval is only one step toward each developer’s ultimate goal of a cell therapy that is sustainable, scalable, consistently high in quality, and manufacturable with a reasonable cost of goods (CoG).
Companies must take a comprehensive, proactive approach to commercial deliverability. They need practical strategies for defining the characteristics and goals of a product, maintaining a consistent quality plan to achieve a pharmaceutical quality system, creating a strategic plan for commercial-scale manufacturing, and engineering automated solutions that effectively evolve the manufacturing facility beyond the cleanroom into the “factory of the future.” Below, we offer our perspective on each of those critical success factors.
The Four Pillars:
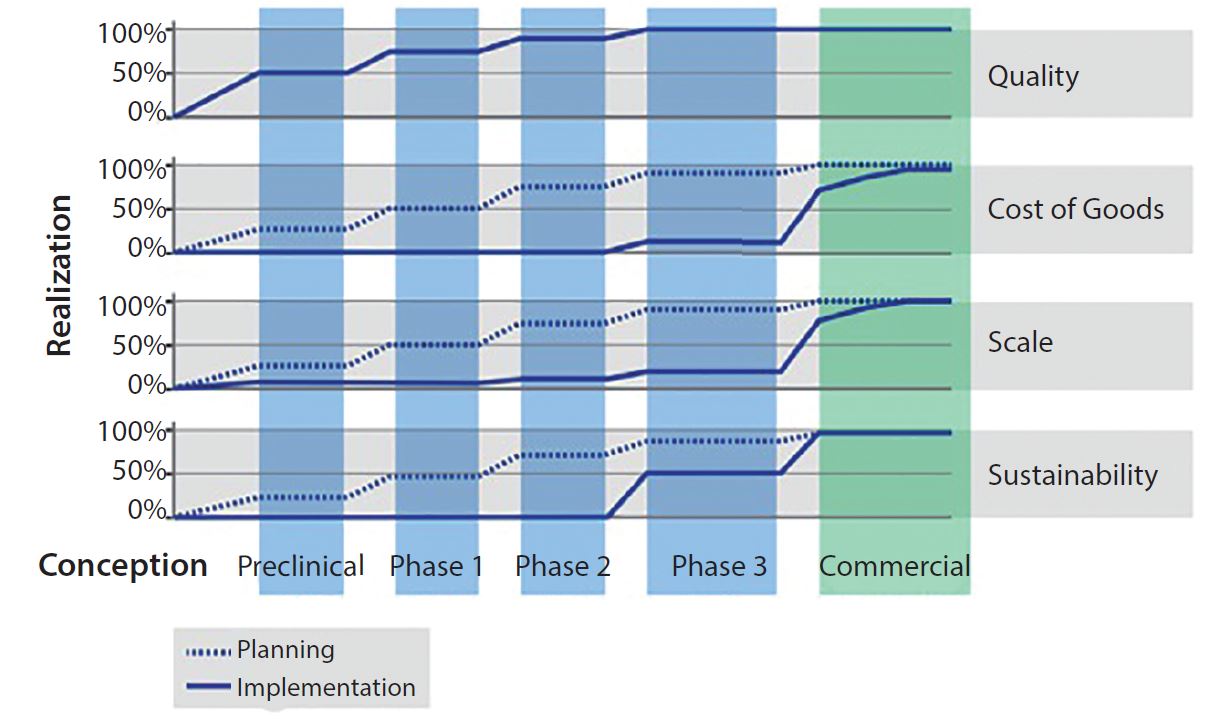
Figure 1: Development by design, initiate early
Development By Design
Development by design (DbD) is a commercial manufacturing strategy that takes into account quality, CoG, scalability, and sustainability. Proactively implementing a DbD strategy does not force a cell therapy developer to make a large investment early in a project, but it does mean that the company must plan ahead. Working through the four facets of DbD (outlined below) at an early stage (including building quality into the process at the onset) can provide significant cost and time advantages as a cell therapy moves along in its clinical process (Figure 1).
Quality is the foundational element of DbD. Cell therapy manufacturers rely heavily on process compliance for critical quality attributes (CQAs) of final products. Manual, open, and human-dependent process steps present substantial risks of contamination. Automation, integration, closed-system design, and compliance with applicable regulatory standards such as current good manufacturing practices (CGMPs) are key tactics to improve control and robustness of processes.
Cost of Goods: Current high CoG for cell therapy products usually demand a sizable commercial value proposition. As processes mature, the focus on CoG for commercial viability becomes critical. Tactics that lower CoG often affect other DbD drivers. For example, implementing closed, automated systems not only reduces the labor component of CoG, but it also addresses quality needs for an error-free and repeatable process. However, a new system can present sustainability challenges if it is a proprietary, sole-source technology from a young company.
Scalability: Migrating from a clinical-scale process (with the capacity to make tens to hundreds of patient doses per year) to a commercial-scale process (with the capacity to make thousands to tens of thousands of patient doses per year) can present significant comparability risks. Incoming raw biological material introduces variability. And new quality risks and other technical challenges often present themselves after a drastic change in scale from clinical to commercial manufacturing.
Sustainability: Even if quality, CoG, and scale objectives have been met, cell therapy developers face a very real risk that manufacturing cannot be sustained over a product’s full life cycle. Limitations on critical raw material availability caused by single-source supply, fluctuating availability of dedicated manufacturing facilities and personnel, and reimbursement challenges are just a few potential roadblocks to sustainability. To mitigate such risks, companies must assess their full range of supply-chain inputs to their manufacturing processes. Those include reagents, consumables, equipment, and human resources. Assessment should methodically include every unit operation in both manufacturing and testing.
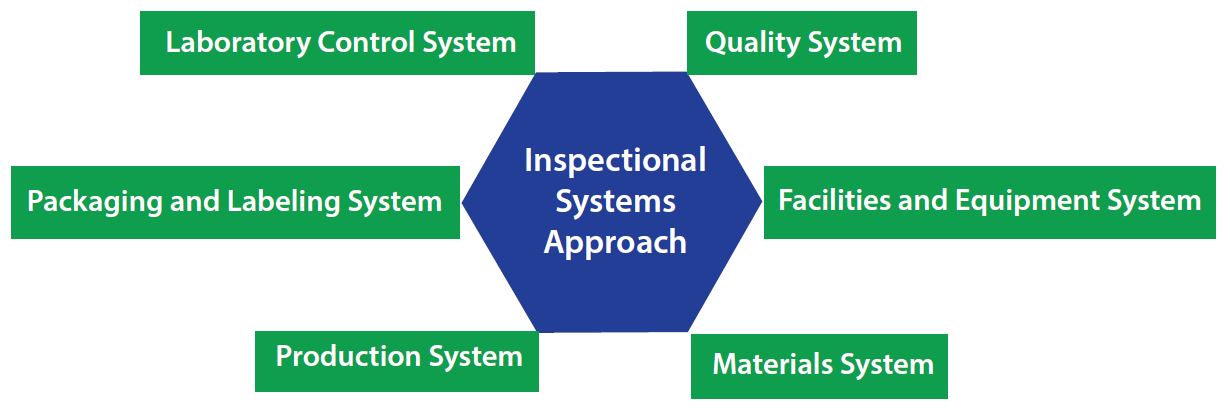
Figure 2: Integrational systems approach
Good Quality Is Good Business
For a cell therapy developer, one of the most important (and most complex) requirements for scaling up to commercialization is meeting quality standards, including compliance with applicable regulations such as CGMPs. Implementing a quality plan is desirable for achieving FDA-approvable commercial manufacturing readiness, whether you work in a dedicated facility designed solely for your own therapies or with a manufacturing partner. A quality plan should incorporate an inspectional-systems approach that takes into account the following systems: quality, facilities and equipment, materials, production, packaging and labeling, and laboratory control (Figure 2).
A manufacturing partner that is truly prepared for commercialization (particularly in a shared manufacturing facility) will exhibit a foundational state of compliance underpinning the “right to operate” in a compliant CGMP manufacturing environment. That includes consistent adherence to your own quality systems consistent with applicable regulatory requirements. Such requirements include CGMPs, both within a manufacturing site and across interactions with all developers that partner with that site. Although flexibility to meet developers’ needs may seem advantageous, a manufacturer that changes basic quality systems and processes for every developer opens itself up to potential risks related to product comparability and nonadherence to applicable quality regulations. A unified, consistent, foundational state of compliance that is harmonized with best industry practices is the best way to mitigate potential regulatory risk.
Cell therapy developers should establish at their manufacturing facilities (or look for manufacturing partners that offer) that foundational state of compliance in people, facilities, systems, and processes, as outlined below.
People: Establish a facilitywide, multidisciplinary quality task force to lead parallel work streams that address the quality requirements for commercial manufacturing.
Facilities: Implement facility design and renovations to ensure a strong foundational state of CGMP compliance that will support drug development strategies, incorporate on-site commercial manufacturing capacity, fulfill critical commitments to cell therapy developers, and close identified compliance gaps.
Systems: Unify, align, and integrate harmonized quality systems across a shared facility that will withstand regulatory scrutiny, including implementation of electronic quality systems where possible.
Processes: Establish a seamless technology transfer process (the transfer of technology information from a cell therapy developer to a shared facility) that allows for commercial product manufacturing.
Once a unified quality voice is established, a manufacturer must determine which quality elements contribute to the successful transition from clinical to commercial readiness. Those elements include process comparability and all quality metrics that demonstrate comparability, control (including appropriate process and quality controls to ensure patient safety), compliance with all applicable regulatory standards such as CGMPs, and consistency of all quality metrics used to assess quality systems and processes (such as timelines, system checkpoints, and documentation procedures).
Finally, a best practice for a manufacturing partner to achieve commercial-readiness from a quality perspective is to establish a quality task force (QTF). This team should include representatives from manufacturing, operations, and quality groups, with different areas of industry experience. The purpose of a QTF is to proactively lead a manufacturing facility and mobilize multidisciplinary resources toward establishment of proven commercial quality systems. Such systems should withstand regulatory scrutiny and meet all applicable quality standards and regulatory requirements.
To understand the value of a QTF, consider the fact that many cell therapy developers that operate dedicated manufacturing facilities will bring in outside expertise when they need to optimize or overcome challenges related to quality. This expertise often is not sought out until a developer is in the troubleshooting (or worse, rescue-mission) stage after quality oversights or hurdles have resulted in product development delays, regulatory actions, or standstills. A QTF incorporates what you have learned and your experience so that it can be applied across all developers as a best practice moving forward.
Ultimately, a quality division’s primary concern is patient safety. Without adherence to quality standards, a cell therapy process cannot meet regulatory scrutiny and produce a commercial product.
Know Thyself: Defining a Cell Therapy Product
The FDA has provided draft guidance for creating a target product profile (TPP) (for biologics as well as cell therapies) to document product attributes in a format that can evolve into label claims for a product. Cell therapy developers should create a TPP (or similar approach) to guide product development.
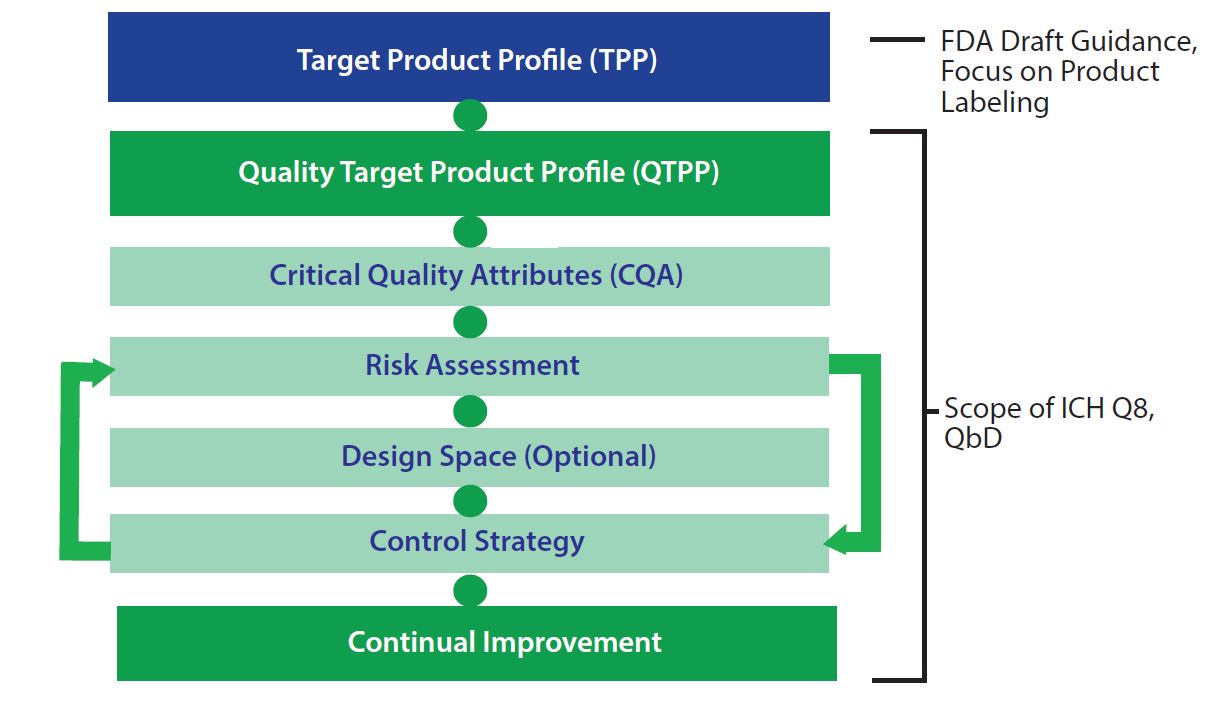
Figure 3: Element of the target product profile and quality by design
The FDA has also provided guidance from the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) Q8 document for pharmaceutical development (where QbD principles are presented) for establishing a quality target product profile (QTPP) (1). A QTPP document typically includes critical quality attributes (CQAs), risk assessment, a design-space plan, and a control strategy (Figure 3). Using a TPP as a key source of input, manufacturers can create a QTPP and used it to guide development. That typically includes detailed targeted postmanufacturing product attributes needed to support a product’s safety and efficacy.
Despite some latitude in how a QTPP is constructed, a recommended format includes
- characteristics profile (e.g., description, formulation, dosage, potency, volume, and shelf life)
- safety profile (e.g., microbial assurance, cellular impurities, and manufacturing residuals)
- use profile (e.g., indications for use, treatment timing, preparation, and use)
- business profile (e.g., geographies, market projections, clinical/ commercial milestones, CoG targets, and intellectual property).
Often when cell-therapy developers begin clinical development of a product, many elements of a QTPP (e.g., specifics of the use profile and business profile) are not yet fully known. So the tendency is to postpone drafting a QTPP until most of the necessary information can be specified. Unfortunately, that approach often leads to questionable manufacturing development decisions that may compromise an overall program. For example, postponing development of a QTPP can lead to cost overruns, increased comparability risks, and timeline delays. A QTPP should be developed (at least as an initial draft for guidance) during early phases of clinical development.
When developed early, a QTPP provides the framework for a rational development plan that starts with the end in mind. The plan then can be used to maintain strategic alignment as a therapeutic program develops and to manage risks inherent to cell-therapy manufacturing. When a cell-therapy developer is ready to engage external resources (such as a manufacturing partner), then this strategic alignment helps to ensure that both developer and manufacturer start from the most informed perspective possible.
Customized Roadmap: a Strategic Commercial Manufacturing Plan
With DbD and quality in mind, a prudent cell therapy developer will initiate and produce a strategic commercial manufacturing plan (SCMP) through either its own efforts or (more likely) with the support of an experienced manufacturing partner. To ensure that a developer’s manufacturing process takes into account the four facets of DbD, an SCMP document is intended to provide a comprehensive, strategic, detailed roadmap to commercial deliverability. An SCMP incorporates elements from a strategic manufacturing assessment (SMA) and quality risk analysis (QRA) and uses them among other building blocks to deliver a thorough assessment and to provide a clear path to commercial success.
An SCMP should include three primary segments: an evaluation of a developer’s current manufacturing processes; an analysis of those processes for areas of potential optimization and improvement; and a practical, implementable strategy to take a process from its current state to a future, commercial-ready state while reducing risks.
The evaluation segment should provide a thorough description of the current state of a developer’s manufacturing process. It should include current-state descriptions of QTPP, critical quality attributes (CQAs), and unit operations.
In the analysis segment, a manufacturing partner considers the information derived from the evaluation segment. That information helps identify opportunities for optimization and improvement as well as particular areas of challenge and risk. Here, strengths and weaknesses in a developer’s manufacturing process are identified to outline what is needed to successfully evolve from the current state to a state of commercially viable manufacturing. Manufacturers must determine whether anything about the current state of their products and processes might lead to problems later in clinical development. Does a product pose a comparability risk or have too high a CoG? Is it not scalable to meet market demand or not sustainable over its commercial life?
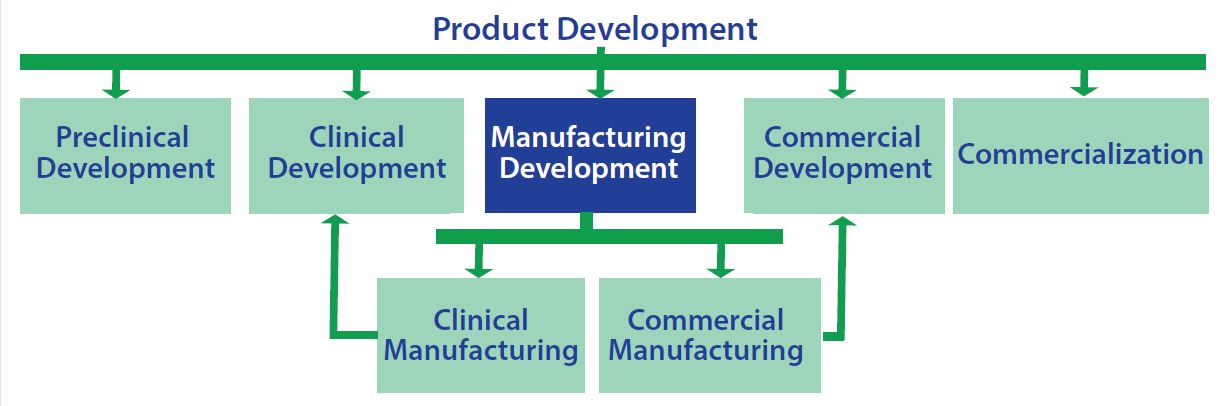
Figure 4: Although the scope of a strategic commercial manufacturing plan is focused on manufacturing development, the plan must be crafted and executed in the context of all the other elements of product development
Strategy: Finally, an SCMP provides concrete recommendations in the form of an optimization plan based on evaluations and analyses performed. Successful implementation of this plan is needed to meet commercial manufacturing. Although the scope of a SCMP is focused on manufacturing development, the plan must be crafted and executed in the context of all the other elements of product development (Figure 4).
Factory of the Future
In the history of cell therapy manufacturing so far, many developers have invested substantial time and resources creating facilities dedicated solely to the manufacture of their own products. Operating costs, difficulty scaling up/out or down to meet market demand, and an inability to predict labor intensity and staffing needs in such facilities too often have proven to be insurmountable obstacles. Such difficulties have led to idle capacity: underused technologies, people, facilities, and resources. Providing a steady flow of scalable, automated, high-volume, mass-produced product is the only way to effectively distribute costs and risks.
The most logical solution to this industrywide problem — and a way for the industry as a whole to advance — is for cell therapy developers to adopt a “shared risk” approach to their manufacturing facilities. This strategy, allows for the spread of idle capacity among a greater number of developers through the use of manufacturing partners, thereby reducing risk for all. It is prudent for cell-therapy developers to follow the example set by the pharmaceutical industry in leveraging shared-risk facilities.
One successful approach has been to use a combination of dedicated facilities for small-scale or early clinical work and move to manufacturing partners as scale increases and commercialization approaches. Although manufacturing for clinical development involves a great deal of scale-up or scale-back to meet market demand, once commercial development begins, a facility needs to have the capacity to continue producing high volumes of products consistently. That way, cell therapy developers can ensure that a manufacturing facility itself does not hinder commercial viability. Concentrating world-class expertise and a unified quality system at a shared facility are important added benefits.
However, simply building a better and bigger clinical manufacturing infrastructure is not enough for successful commercial manufacturing. To overcome challenges outlined herein, the industry needs a comprehensive effort of innovation and engineering to rebuild unit operations for cell therapy manufacturing from the ground up. Instead of performing cell therapy manufacturing in designated facilities (or even designated cleanrooms), the industry must focus on innovation, engineering, and automaton to create closed-processing systems. Such systems allow for unit operations to be entirely self-contained within manufacturing equipment and testing instruments. A few such systems are already in development and beginning to reach the market. Examples include the CliniMACS Prodigy automated cell-processing system from Miltenyi Biotech, the Integrated Cell Processing Work Station from Panasonic Healthcare, the Octane Cocoon system, and an upcoming technology that we (through PCT) are developing in partnership with Invetech.
If cell therapy manufacturers adopt such systems and the closed model of manufacturing, then their work could be performed outside cleanrooms. Instead, it can happen in spaces where multiple pieces of equipment can manufacture multiple therapies in close physical proximity. Only when manufacturing facilities complete their transformation to such “factories of the future” — those more suited to high-volume production — can the cell therapy industry (specifically the manufacture of patient-specific therapies) achieve commercial scalability.
Looking into the Crystal Ball: The Way Forward
Cell therapies offer enormous promise for patients in need of new treatment options. These technologies have delivered amazing results in challenging therapeutic areas such as cancer. For individual therapies and the industry as a whole to advance toward and thrive in a commercial state, product developers and their manufacturing partners must focus on deliverability. A number of practical, yet easily overlooked strategies are essential to put in place:
- Define a cell therapy product with a quality target product profile.
- Proactively plan for future clinical phases with the four facets of DbD (quality, cost of goods, scalability and sustainability).
- Maintain a consistent quality plan and foundational state of compliance.
- Initiate a strategic commercial manufacturing plan to assess the current state of commercial readiness and achieve actionable solutions to manufacturing challenges.
- Optimize the facility design itself to evolve beyond cleanrooms. The type of proactive, strategic approach described here will help developers overcome the inherent difficulties of cell-based therapeutics and make steady progress toward successful commercial-scale manufacturing.
Reference
1 Guidance for Industry: ICH Q8(R2): Pharmaceutical Development. US Food and Drug Administration: Rockville, MD, November 2009.
Corresponding author Bob Preti, PhD, is president of PCT and senior vice president of development and technical operations and chief technology officer at Caladrius Biosciences. Ann M. Daus, PhD, is vice president of quality assurance, quality control, and validation at PCT. Brian Hampson is vice president of global manufacturing sciences and technology (MSAT), and head of the Center for Innovation and Engineering. Cenk Sumen is manager of technology and business development at PCT; 1-201‐883‐5300; www. pctcaladrius.com.