
www.photos.com
Generating a stable environment for a biopharmaceutical drug substance is a critical step for ensuring a long drug-product shelf life (1–6). This process begins early in development with preformulation screening. Some of the most critical parameters to maintaining potency and activity are protein conformation (tertiary or three-dimensional (3-D) structure), folding (secondary structure), and proper subunit association (quaternary structure). Collectively, those are known as higher-order structure (HOS) and can be highly influenced by the formulation environment of a protein drug product.
But analytical monitoring of protein conformation is frequently omitted from preformulation because of time constraints, the complexity of most common techniques, and the inherent lack of sensitivity of some biophysical methods to subtle structural changes. However, combining traditional biophysical techniques with more sensitive orthogonal approaches can provide a comprehensive view of HOS in biopharmaceutical formulation.
Biophysical analysis has become increasingly important in the characterization of biopharmaceutical drug candidates. Such methods may be described as covering a mixture of disciplines, being essentially the use of physical techniques to investigate and characterize biological systems (7). The wide range of technologies within this area for elucidating 3-D protein structure includes spectroscopic, thermodynamic, and hydrodynamic techniques such as circular dichroism (CD) (8–11), Fourier-transform infrared (FTIR)(12–15) spectroscopy, differential scanning calorimetry (DSC), intrinsic and extrinsic fluorescence (16, 17), dynamic light scattering (DLS), and sedimentation velocity analytical ultracentrifugation (SV-AUC) (18–20). Such analyses can provide valuable information about secondary, tertiary, and quaternary structures of protein molecules (e.g., monoclonal antibodies) and provide evidence of their conformational stability.
Biophysical analyses have particular application in determining and confirming the stability and conformation of protein structures. They also can be used to define optimum buffer conditions and characterize degradation pathways and products. So biophysical analysis provides valuable information about how a formulation environment influences stability of a protein molecule (21–25). These techniques also provide information that can be used for comparability analysis.
The Role of Biophysical Analysis in Product Development
The emergence and rapid growth of the biosimilars market is driving growing integration of biophysical analyses into biomolecular characterization (22, 23). About 15 years ago, the biopharmaceutical industry was somewhat skeptical about biophysical analysis. Although it was considered to be state-of-the-art technology, widespread acceptance of biophysical methods was hindered by a lack of compliance and streamlined purpose to fulfill regulatory and practical requirements of the biopharmaceutical industry (e.g., regarding sample consumption in early development). Biophysical analysis was seen as an academic discipline to be applied only in strict research and development (R&D) environments. Consequently, its use remained underdeveloped in the biopharmaceutical sector.
The emergence of biosimilars changed that status quo. Biosimilar drug manufacturers needing to provide technical comparability data found biophysical analysis to be a valuable analytical tool. Visionary contract research organizations (CROs) such as Avacta Analytical and SGS M-Scan were early promoters of the use of this type of analysis, but they faced a challenge in persuading customers of its true value. Generally, larger companies with extensive testing capabilities already were fully committed to using such techniques for protein characterization. But most medium-to-small biopharmaceutical companies have neither the capacity nor the internal expertise to implement biophysical analysis. Unfortunately, the need for HOS characterization often was widely ignored at many levels. Funds needed to invest in the required technology were neither available nor justifiable by those companies. Furthermore, 1999 regulatory guidelines (ICH Topic Q6B) did not sufficiently emphasize the relevance of biophysical analysis — nor do they today — and challenges associated with collection and productive interpretation of the so-called spectroscopic profiles.
In addition to that ominous picture, lack of a competitive CRO market providing biophysical services perpetuated that vicious cycle until the arrival of biosimilars. Together with technological advances in instrumentation, market education on HOS was part of this recurring issue. Over recent years, that less-than-bright picture has improved substantially, with gradual inclusion of biophysical instrumentation as part of routine analyses used for biopharmaceutical characterization. As a response to a greater presence of CROs active in this area, such instrumentation has become more streamlined to current industry requirements. Meanwhile, regulatory guidelines are becoming more proactive in emphasizing the importance of protein HOS analysis. Perhaps more important, an increasingly demanding market and more educated industry is the force behind these deterministic changes.
A Case Study
Currently, FTIR and CD are the most commonly used methods for determining protein structural conformation. Both can be insensitive to subtle tertiary conformational changes that may be caused by small differences in the formulation environment. Below are data obtained from analyzing three different formulations of bovine immunoglobulin G (IgG1) in an attempt to reduce the electrostatic dimeric content of IgG. SV-AUC data indicate that one formulation successfully mitigated oligomerization of bovine IgG but induced small changes in its thermal stability and surface hydrophobicity area.
Materials and Methods: My company tested three different formulations (A, B, and C) of bovine IgG at t0 days with a selection of biophysical methods: a Beckman Coulter XL-A AUC instrument, a Jasco J-815 CD spectropolarimeter for near and far CD, a Bruker Tensor 27 spectroscope equipped with AquaSpec and Bio-ATR accessories for FTIR, a Shimadzu RF5301 spectrofluorometer for steady-state intrinsic and extrinsic fluorescence, and a Setaram μDSC 3EVO instrument for DSC. Additionally, we incubated bovine IgG in formulation C at 2–8 °C at different time points up to 45 days (t15, t30, and t45 days) before analysis by near and far UV CD and intrinsic steady-state fluorescence. Lyophilized bovine IgG was dissolved in the respective formulation buffers at concentrations of 1 mg/mL (SV-AUC, CD, and intrinsic fluorescence); 10 mg/mL (FTIR, DSC); and 20, 40, 80, and 100 μg/mL for extrinsic fluorescence experiments.
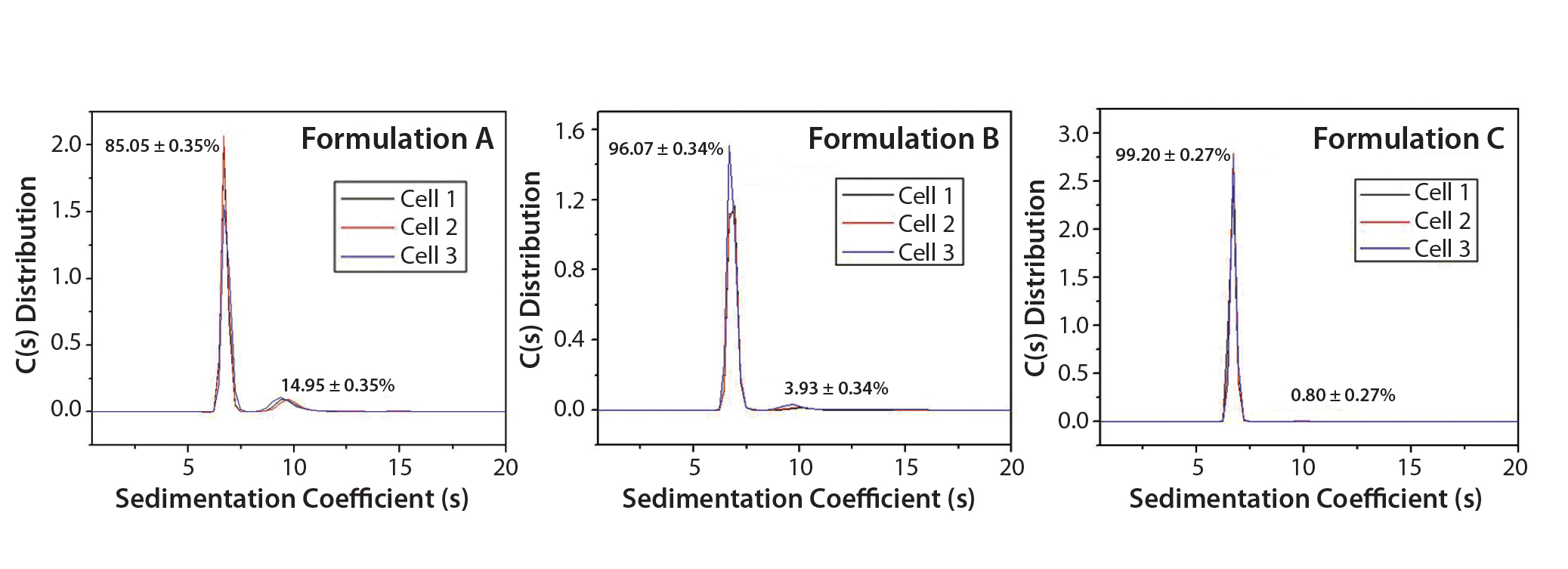
Figure 1: SV-AUC analysis of IgG in formulation buffers A, B, and C
Results: SV-AUC results (Figure 1) showed evidence of dimerization in bovine IgG at t0 in formulation buffer A. By contrast, formulations B and C at t0 showed less dimerization (Figure 1, Table 1). SV-AUC measurements indicated that both B and C were successful in mitigating the electrostatic interactions that led to formation of dimeric species.
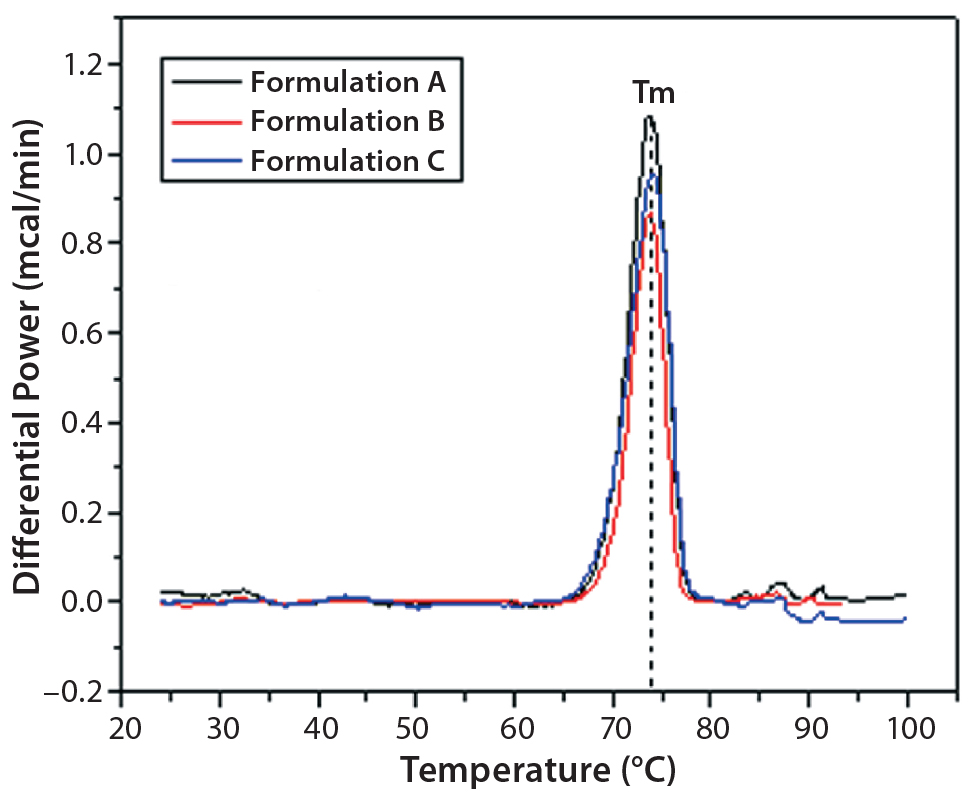
Figure 2: DSC traces of IgG in formulations A, B, and C
DSC measurements (Figure 2, Table 2) showed identical melting points for formulation A and B (73.87 °C and 73.63 °C, respectively), detecting only small differences for formulation C (73.01 °C), which may indicate less thermal stability for bovine IgG in formulation C. Extrinsic fluorescence measurements confirmed that view. Figures 3 and 4 show a small but different linear response in the fluorescence emission spectra of bis-ANS as a function of concentration of IgG in formulation C relative to formulation A and B. CD (Figures 5 and 6, Table 3), FTIR (Figure 7, Table 4), and intrinsic fluorescence measurements (Figure 8) aiming to elucidate the secondary and tertiary structure of proteins showed that the different composition of formulations A, B, and C did not affect the structure of bovine IgG.
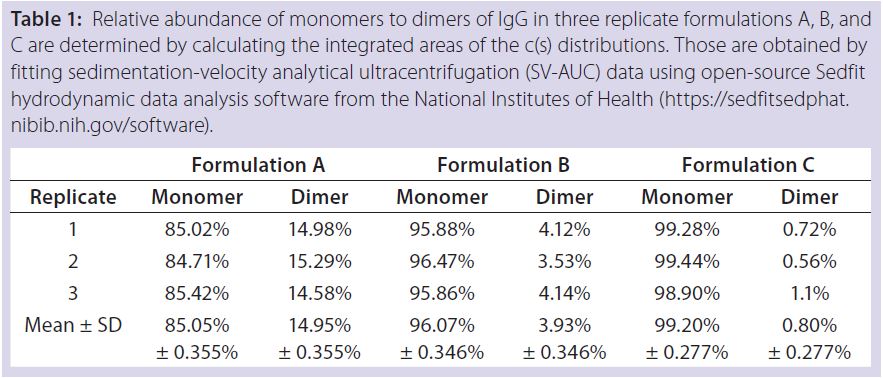
Table 1: Relative abundance of monomers to dimers of IgG in three replicate formulations A, B, and C are determined by calculating the integrated areas of the c(s) distributions. Those are obtained by fitting sedimentation-velocity analytical ultracentrifugation (SV-AUC) data using open-source Sedfit hydrodynamic data analysis software from the National Institutes of Health (https://sedfitsedphat.nibib.nih.gov/software).
Figures 9 and 10 show far and near UV CD data accumulated for bovine IgG at t0, t15, t30, and t45 in formulation C. Those data showed a loss of ellipticity in the far UV from t30. Changes in tertiary structure were identified at t15 in near UV. Significant changes in the fluorescence spectra of bovine IgG in formulation C were detected as early as t15 days. From t0 to t45, a progressive increase in fluorescence emission quantum yield and blue shift of the emission spectra was observed (Figure 11).
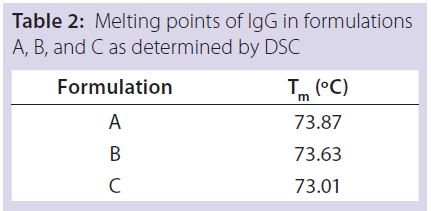
Table 2: Melting points of IgG in formulations
A, B, and C as determined by DSC
Conclusions: Commonly used FTIR and CD methods for determining protein structural conformation can be insensitive to subtle tertiary conformational changes induced by small changes in the formulation environment. Data obtained from analyzing three different formulations of bovine IgG1 showed that formulation C was successful in mitigating the oligomerization of bovine IgG at the expense of inducing small changes in its thermal stability and surface hydrophobicity area. Secondary and tertiary structure appeared unaffected as reported by CD, FTIR, and steady-state fluorescence. The minor differences detected on thermal stability and exposure of hydrophobic residues on the surface of bovine IgG are understood to be induced by subtle changes in the ensemble tertiary structure of IgG as a response to its formulation in buffer C. Our orthogonal use of biophysical methods prompted a more detailed investigation of the refrigerated stability capabilities of bovine IgG in formulation C.
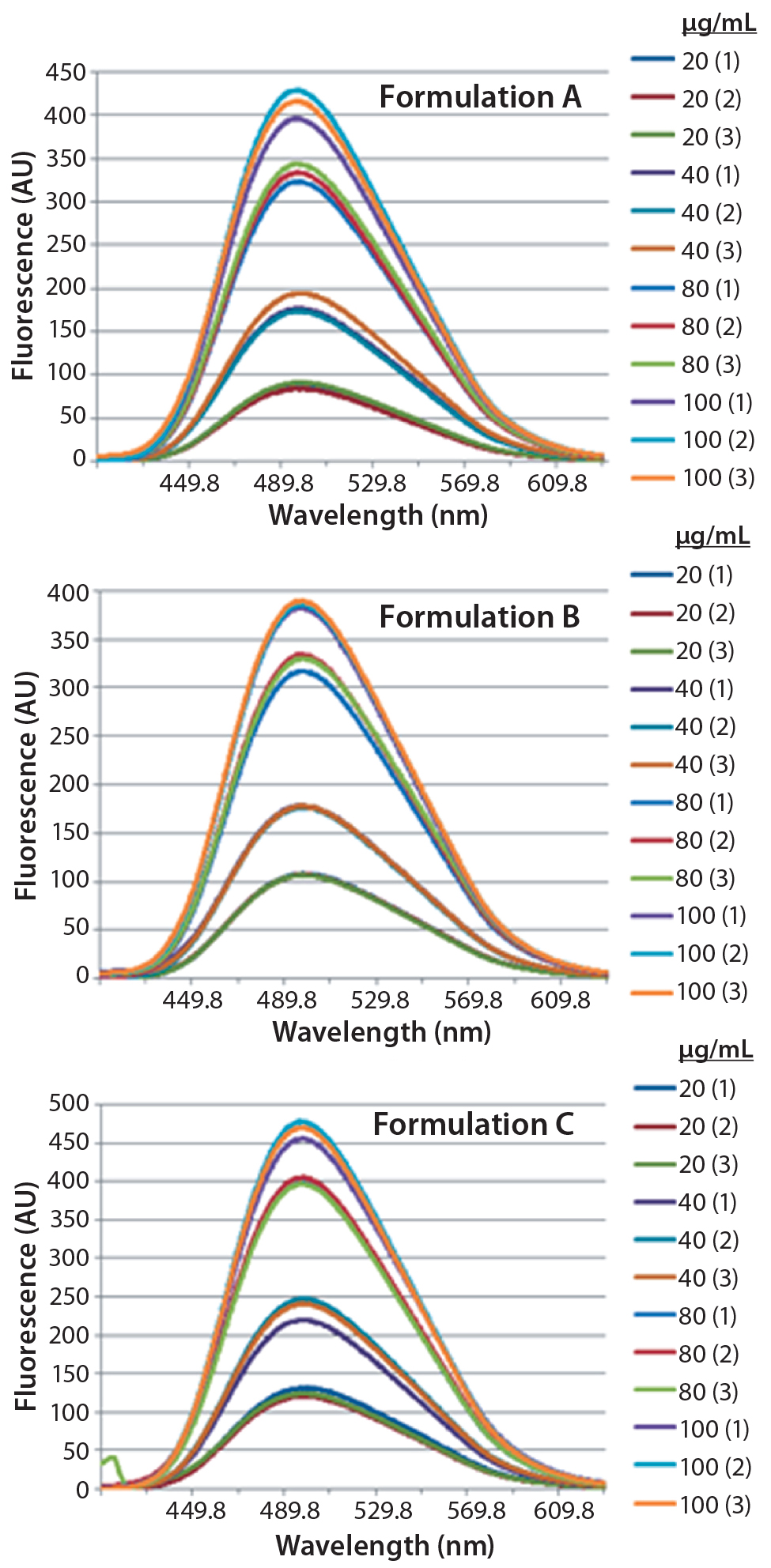
Figure 3: Steady-state extrinsic fluorescence emission spectra of bis-ANS (10 μM) in the presence of increased concentrations of bovine IgG (20, 40, 80, and 100 μg/mL) in formulation A (upper panel), formulation B (middle panel), and formulation C (lower panel), respectively
Resulting refrigerated stability data showed that bovine IgG formulated in that buffer system had a propensity to lose tertiary and secondary structure along set time points that were not seen with formulations A and B (data not shown). Those results confirmed our view that formulation C was a successful buffer system to reduce electrostatic dimerization of bovine IgG but that it induced subtle tertiary structural changes at t0. Those changes, by exposure of an increased hydrophobic surface area, were critical to the refrigerated conformational stability of bovine IgG in that specific formulation.
These data demonstrate the importance of selecting orthogonal stability-indicating methods for conformational stability in formulation studies. Key biophysical markers of stability can be overviewed easily or omitted otherwise. Minor conformational changes can expose regions of a protein molecule that are normally protected within its internal structure, potentially leading to unexpected degradation.
Choice of Technique: Experimental Strategy
The choice of biophysical techniques to use for protein structural analysis depends on whether a study’s objective is to characterize the biomolecule (typically the case for new biopharmaceutical entities) or provide comparability data (the usual scenario in characterizing biosimilars). Either way, the aim is always to optimize a strategy in relation to the intended purpose of the study.
The difficulty lies in that all biophysical methods have their limitations, so no single technique provides all required information. For example, SV-AUC provides accurate quantification of aggregates but gives only an estimate for the molecular weight of a protein. Thus, size-exclusion chromatography with multiangle laser light scattering (SEC-MALS) should be used as an orthogonal complement to any SEC platform with single detection and SV-AUC for a direct determination of molecular weights. Similarly, SV-AUC should be used with SEC-MALS (and SEC by extension) for quantification of aggregates.
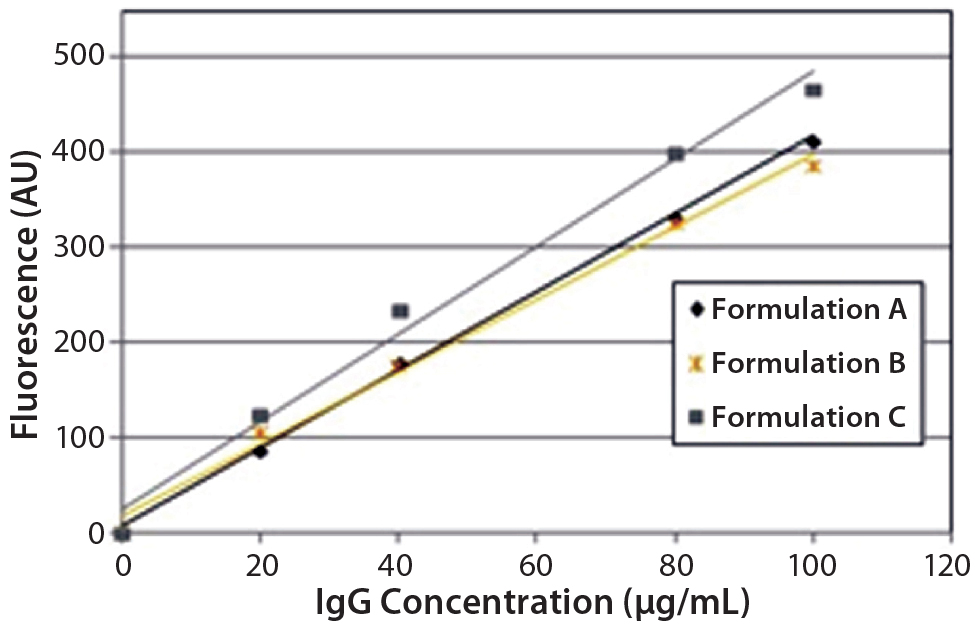
Figure 4: Steady-state extrinsic fluorescence (bis-ANS) measurements; linear response of the fluorescence emission spectra of bis-ANS as a function of concentration of IgG in formulations A, B, and C.
Most researchers tend to believe that a favorite technique is more powerful or versatile than it actually is. So it is important to plan carefully which techniques will be used to optimize the outcome of a study program. Most biophysical techniques should be used in orthogonality. They provide information on the same aspects of a given molecule from different physical perspectives. And because each technique has its own limitations, researchers need to build an “analytical landscape” to appreciate the behavior of a given molecule rather than obtain a biased picture of it by using just one single technique.
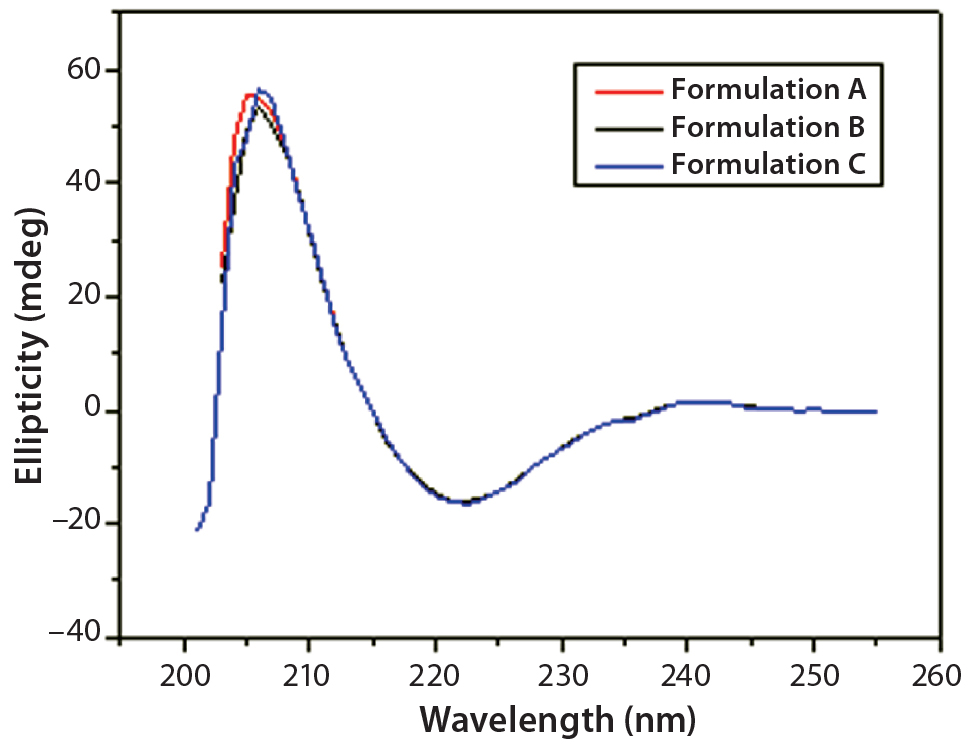
Figure 5: Far-UV CD spectra of IgG in formulations A, B, and C
Build a Collage: Every individual analytical method presents a distortion of reality. Thus, formulators need to build a picture from those pictures — a true analytical collage — to achieve a more accurate conclusion from the test results obtained. If CD is used, for example, some information about secondary and tertiary structures of a protein will be obtained. However, other techniques such as FTIR or fluorescence studies reporting on the same quality attributes of that molecule will provide additional evidence-based information to enable an analyst to reach more broad-based and useful conclusions.
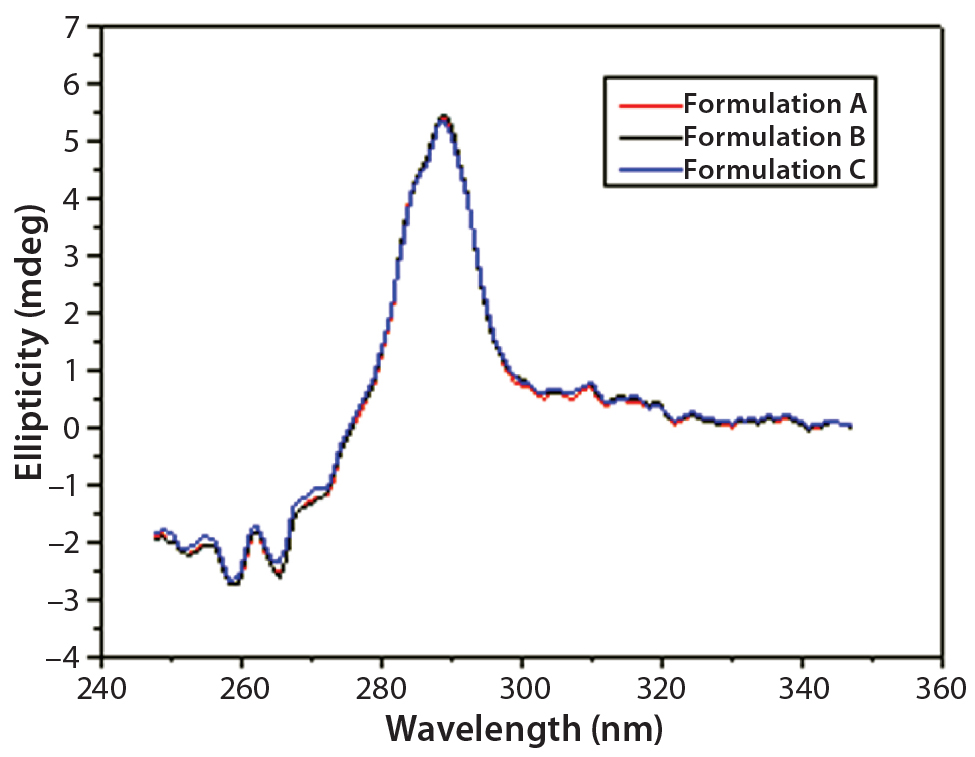
Figure 6: Near-UV CD spectra of IgG in formulations A, B, and C
How should researchers go about choosing the tests to use? This choice is intimately linked to experimental strategy, which in turn depends on project requirements. This is a key consideration. For CROs, it is essential that chosen tests are part of an experimental strategy that satisfies customer needs. And the experimental strategy dictates the choice of technique. Omitting a critical technique from a program leaves a gap in obtained knowledge, which could compromise conclusions drawn from the overall study.
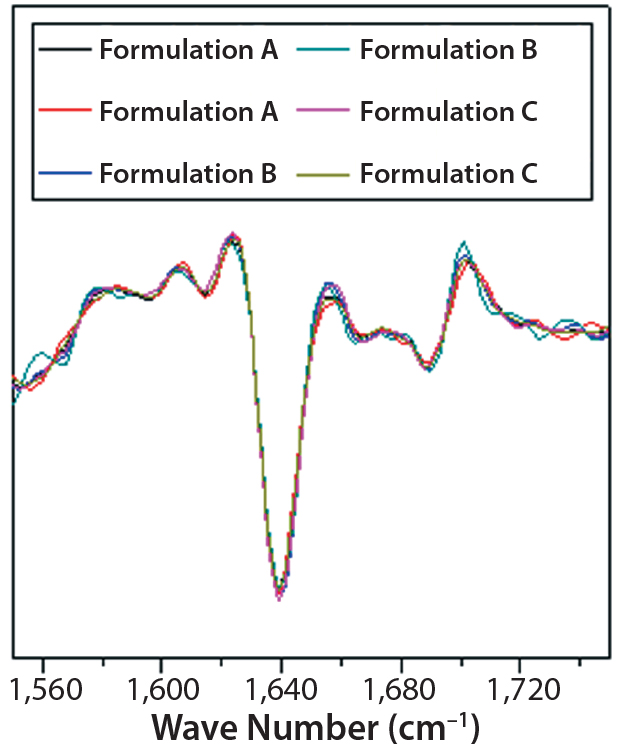
Figure 7: Second-derivative, normalized ATRFTIR spectra of IgG in formulations A, B, and C
Generally speaking, during early characterization studies in biopharmaceutical discovery research, only small amounts of material are available. It is therefore common to use a small number of experiments. Biophysical techniques are considered to be “big expenders” that use large sample amounts. As more material becomes available when development progresses, however, researchers may decide to perform further tests using orthogonal methods to build up the analytical collage for a given molecule. It is important to establish the purpose of those tests because extra work in performing more complex characterization needs to be justified. The amount of work that can be performed depends strongly on the amount of material available.
Comparability studies do not face such strong restrictions on the number of tests that can be performed (within reason) because by the time a comparability study is designed, more material is usually available. My company’s general experience has been that biopharmaceutical customers can supply enough material for such testing purposes, so sponsors are inclined to use several techniques (usually as many as are practically possible).
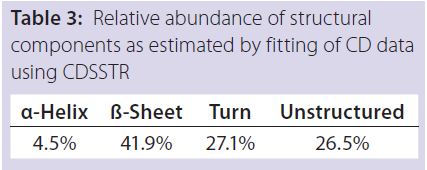
Table 3: Relative abundance of structural
components as estimated by fitting of CD data
using CDSSTR
The object of the study is to provide technical proof of comparability: that a biosimilar product is indistinguishable from the original innovative molecule. In this scenario, it is paramount to put in place feasibility studies that optimize experimental conditions, to define independent replication of experiments for building a solid statistical data set, and to assess the use of orthogonality for covering analytical gaps. Doing so will ensure that an array of techniques with different sensitivities fully corroborates the absence/presence of potential structural differences.

Table 4: Relative abundance of structural
components as estimated by fitting FTIR data
in transmission mode using the OPUS Factor
Analysis program
Biophysical analysis is an ideal approach for performing comparability studies on biosimilars. The qualitative nature of these techniques represents no disadvantage at all: What is required is a comparison of structures. For example, the spectrum of a protein obtained using a technique such as FTIR-ATR provides a “fingerprint” that enables a rapid and simple direct comparison between a biosimilar and its reference molecule, which demonstrates that the technique is valuable.
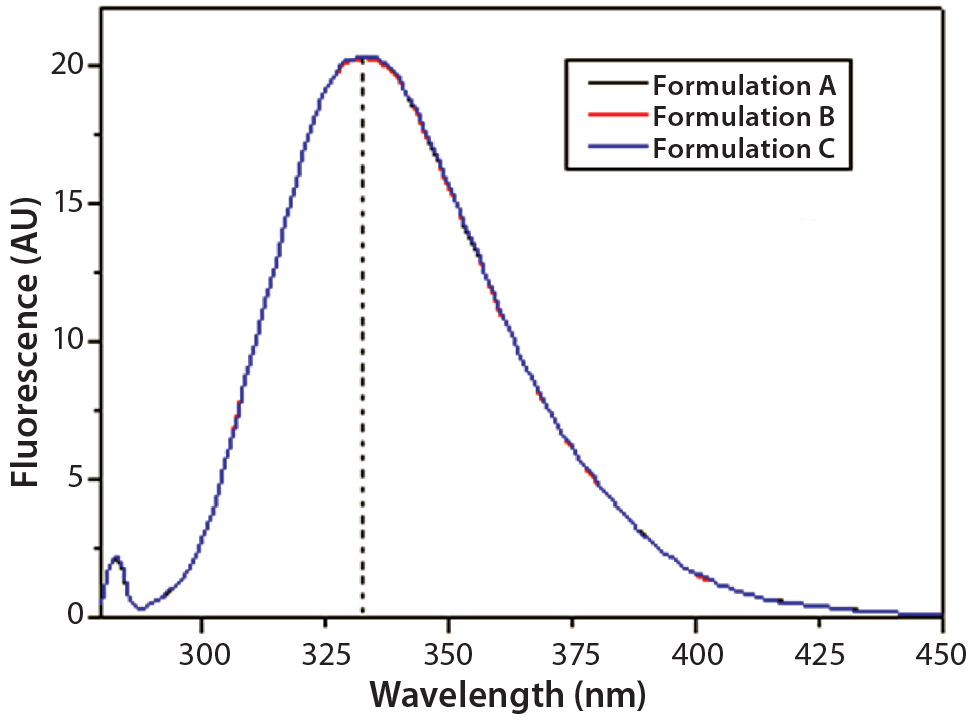
Figure 8: Steady-state intrinsic fluorescence emission spectra of IgG in formulations A, B, and C; the spectrum shown for each formulation is an
average of three independent fluorescence emission spectra collected in three independent sample preparations for each formulation.
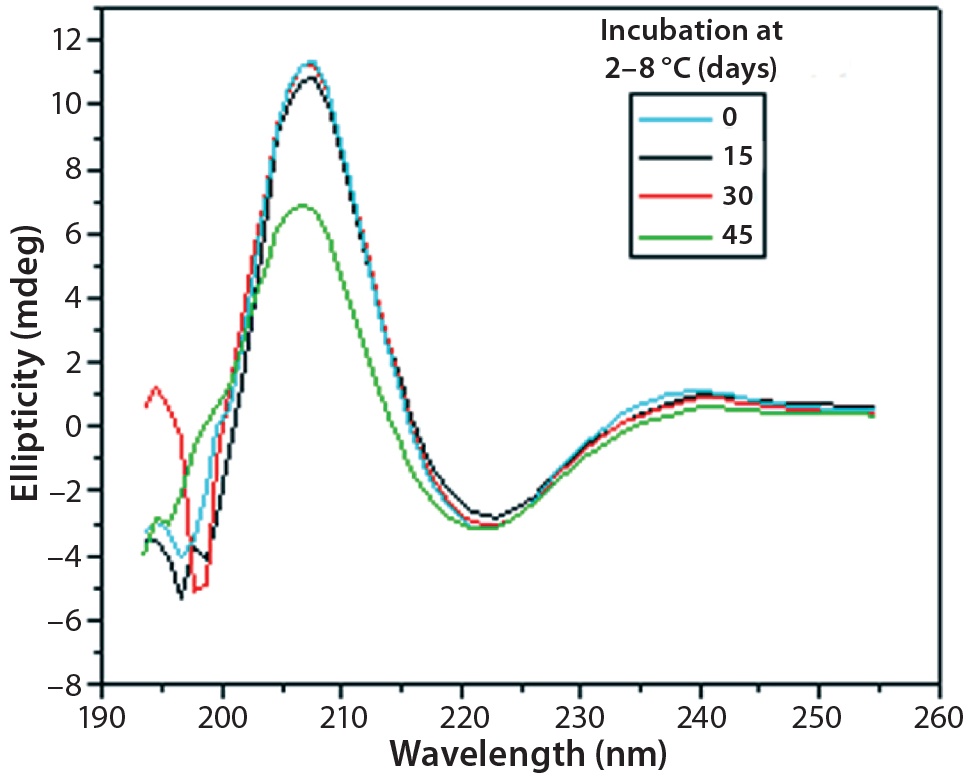
Figure 9: Far-UV CD spectra of IgG in formulation C under incubation at t0, t15, t30, and t45 days
In comparability studies, the experimental approach should be to use as many techniques as possible at any level — quantitative or qualitative — to provide a preponderance of evidence that a biosimilar is truly comparable to its reference molecule. Experimental design is critical. Companies must demonstrate the comparability of biosimilars, including the effects of batch variability. And acceptance criteria for analytical methods used must be defined previous to the comparability study itself.
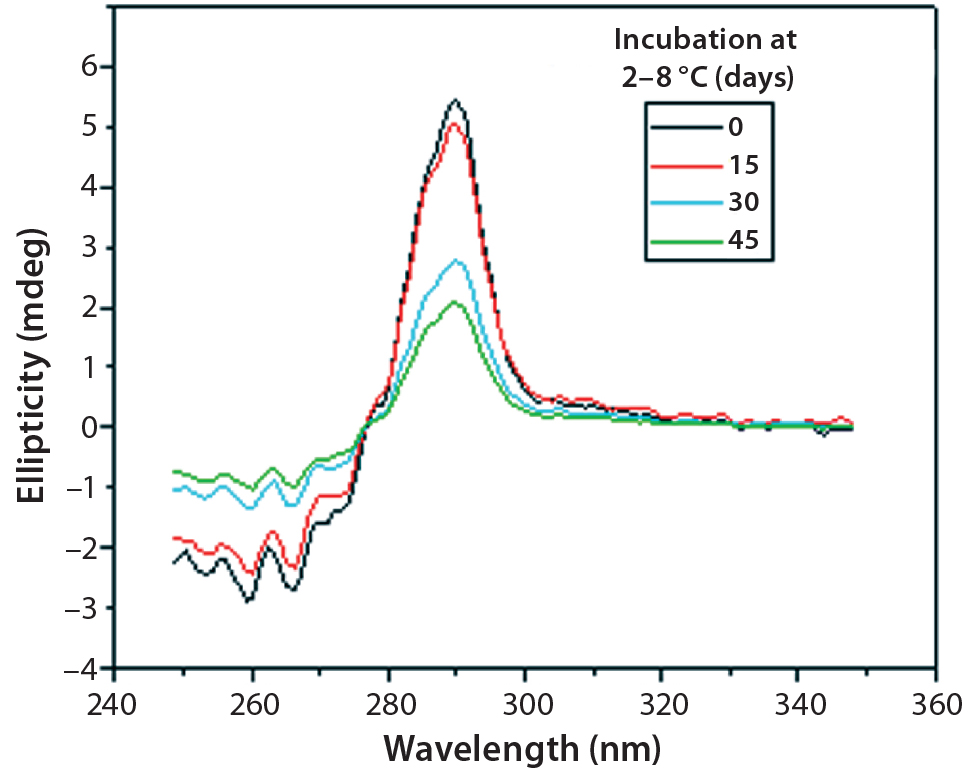
Figure 10: Near-UV CD spectra of IgG in formulation C under incubation
at t 0, t15, t30, and t45 days
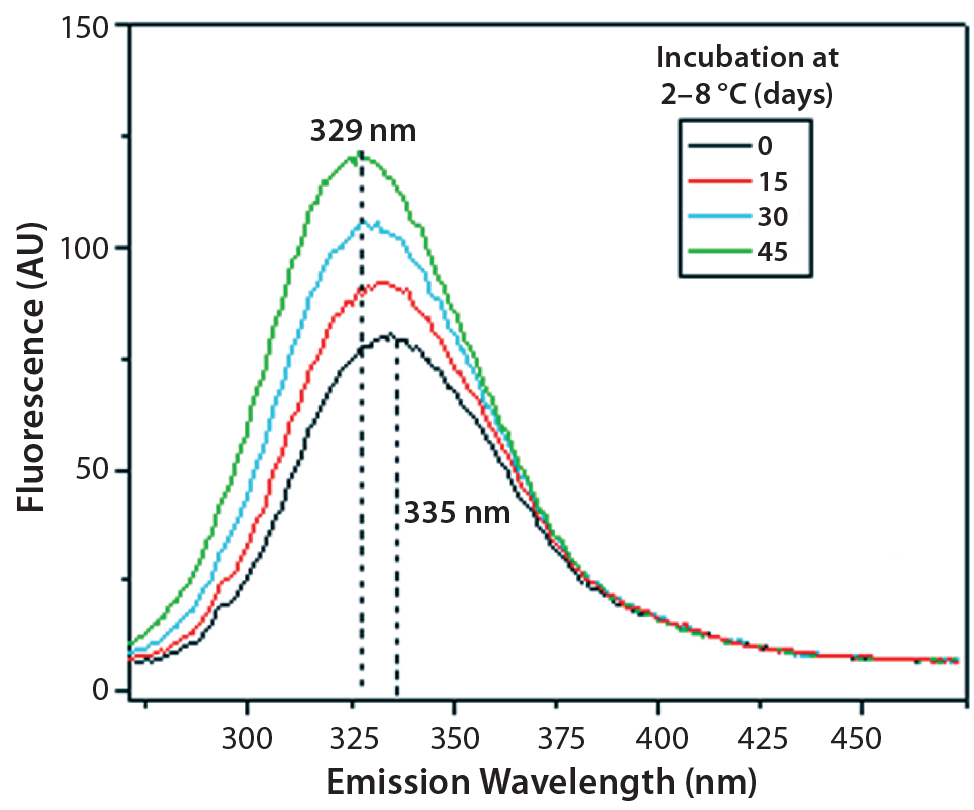
Figure 11: Steady-state fluorescence emission spectra of IgG in formulation C under incubation at t0, t15, t30, and t45 days
A Specialist Service Area
Generally speaking, large biopharmaceutical companies have the capacity and resources to perform their own biophysical analyses, but small and medium-sized companies do not often have the means to fully satisfy the requirements of HOS analysis. Instrumentation cost does not appear to be the main reason, however; data interpretation is a challenge and something of a specialty niche that requires experience and tacit knowledge. The increasingly competitive CRO market supplies many specialty services and technologies. Instrumentation remains something of an issue, as is the education of clients regarding data interpretation.
Over a decade ago, just a handful of service providers offered a broad repertoire of biophysical analysis. Today those numbers are rising to provide a more competitive and diverse service. Challenges remain. Sample consumption is still the Achilles heel of biophysical analysis, which is expensive in terms of sample requirements. Some instrument manufacturers have recognized the need to improve existing analytical platforms to reduce sample consumption and increase sensitivity.
Some interesting developments at a handful of innovative companies are merging the concepts of biophysical and high-throughput analysis. That was a daring fantasy to consider only 15 years ago. New technologies are in development to meet industry requirements as the CRO industry itself is educated about reducing costs and provide maximum value for its biopharmaceutical customers. Biophysical analysis thus is becoming more streamlined to industry requirements. It is no longer simply an academic approach, and the industry is opting in to this rapidly developing area of analysis to support biopharmaceutical product development.
References
1 Formulation, Characterisation, and Stability of Protein Drugs: Case Histories. Pearlman R, Wang YJ, Eds. Springer: New York, NY, 2002.
2 Andya J, et al. Mechanisms of Aggregate Formation and Carbohydrate Excipient Stabilization of Lyophilized Humanized Monoclonal Antibody Formulations. AAPS PharmaSci. 5(2) 2003: 21–31.
3 Kim Y, et al. Effects of Sucrose on Conformational Equilibria and Fluctuations within the Native-State Ensemble of Proteins. Prot. Sci. 12(6) 2003: 1252–1261.
4 Doyle B, et al. Biophysical Signatures of Noncovalent Aggregates Formed By a Glucagon Peptide 1 Analog: A Prototypical Example of Biopharmaceutical Aggregation. J. Pharmaceut. Sci. 94(12) 2005: 2749–2763.
5 Price N. Conformational Issues in the Characterization of Proteins. Biotechnol. Appl. Biochem. 31(1) 2000: 29–40.
6 Wang W, et al. Antibody Structure, Instability, and Formulation. J. Pharmaceut. Sci. 96(1) 2007: 1–26.
7 Chaudhuri R, et al. High- Throughput Biophysical Analysis of Protein Therapeutics to Examine Interrelationships Between Protein Aggregate Formation and Conformational Stability. AAPS J. 16(1) 2014: 48–64.
8 Kelly SM, et al. How to Study Proteins by Circular Dichroism. Biochim. Biophys. Acta 1751(2) 2005: 119–139.
9 Kelly SM, et al. The Use of Circular Dichroism in the Investigation of Protein Structure and Function. Cur. Prot. Pep. Sci. 1(4) 2000: 349–384.
10 Juszczyk P, et al. Circular Dichroism and Aggregation Studies of Amyloid Beta (11-8) Fragments and Its Variants. Acta Biochim. Polon. 52(2) 2005: 425–431.
11 Srisailam S, et al. Influence of Backbone Conformation on Protein Aggregation. J. Am. Chem. Soc. 124(9) 2002: 1884–1888.
12 Kong J, et al. Fourier Transform Infrared Spectroscopic Analysis of Proteins
Secondary Structures. Acta Biochim. Biophys. Sin. 39(8) 2007: 549–559.
13 Harris PI, et al. FTIR Spectroscopic Characterization of Protein Structure in Aqueous and Non-Aqueous Media. J. Molec. Catal. B: Enzym. 7(1–4) 1999: 207–221.
14 Kendrick BS, et al. Quantitation of the Area of Overlap Between Second- Derivative Amide I Infrared Spectra to Determine the Structural Similarity of a Protein in Different States. J. Pharmaceut. Sci. 85(2) 1996: 155–158.
15 Jackson M, et al. The Use and Misuse of FTIR Spectroscopy in the Determination of Protein Structure. Crit. Rev. Biochem. Mol. Biol. 30(2) 1995: 95–120.
16 Royer CA. Approaches to Teaching Fluorescence Spectroscopy. Biophys. J. 68(3) 1995: 1191–1195.
17 Themisttou E, et al. Applications of Fluorescence Spectroscopy to Quantify Shear-Induced Protein Conformational Change. Biophys. J. 97(9) 2009: 2567–2576.
18 Liu J, et al. A Critical Review of Analytical Ultracentrifugation and Field Flow Fractionation Methods for Measuring Protein Aggregation. AAPS J. 8(3) 2006: 580–589.
19 Arakawa T, et al. Aggregation Analysis of Protein Therapeutics. BioProcess Int. 5(4) 2007: 36–47.
20 Wang W, et al. Protein Aggregation and Its Inhibition in Biopharmaceutics. Int. J. Pharm. 289(1–2) 2005: 1–30.
21 Maliket K, et al. Differential Scanning Fluorimetry: Rapid Screening of Formulations That Promote the Stability of Reference Preparations. J. Pharm. Biomed. Anal. 77, 2013: 163–166.
22 Maddux NR, et al. An Improved Version for Multidimensional High- Throughput Preformulation Characterization of Protein Conformational Stability. J. Pharm. Sci. 101(6) 2012: 2017– 2024.
23 Hu L, et al. Investigation of Protein Conformation Stability Employing a Multimodal Spectrometer. Analyt. Chem. 83(24) 2011: 9399–9405.
24 Ahrer K, et al. Thermodynamic Stability and Formation of Aggregates of Human Immunoglobulin G Characterized By Differential Scanning Calorimetry and Dynamic Light Scattering. J. Biochem. Biophys. Meth. 66(1–3) 2006: 73–86.
25 Attri AK, Minton AP. New Methods for Measuring Macromolecular Interactions in Solution Via Static Light Scattering: Basic Methodology and Application to Nonassociating and Self-Associating Proteins. Anal. Biochem. 337(1) 2005: 103– 110. •
Iñigo Rodríguez-Mendieta, PhD, is manager of the biophysical group at SGS Life Sciences Services, 606 Brandywine Parkway, West Chester, PA 19380; 1-610- 696-8210; inigo.rodriguez-mendieta@sgs.com.