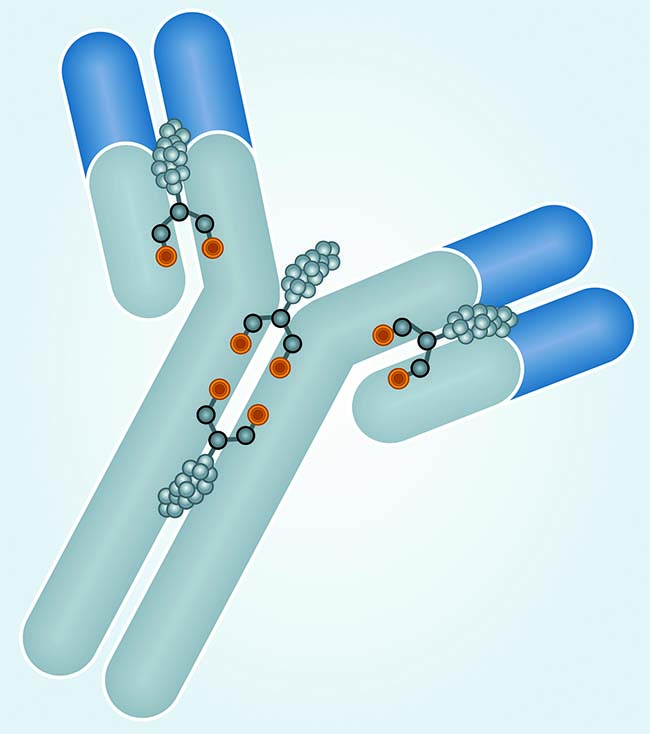
Antibody–drug conjugates (ADCs) for treatment of cancer combine the tumor-targeting properties of antibodies with the cell-killing properties of cytotoxic drugs. By targeting a drug to a tumor, it is possible to reduce systemic toxicity and thereby enable administration of drugs that are otherwise too toxic to be effective therapies. Although the concept of an ADC is simple, in reality developing an effective treatment is somewhat more challenging. Whether an ADC has sufficient efficacy at a tolerable dose depends on four key factors: the target antigen, the antibody, the drug, and the linker between the antibody and the drug.
The Target Antigen
The target antigen should be either specific to or highly expressed on cancer cells (with low levels in normal tissue), not secreted into the circulation and, what is most important, internalized upon antibody binding. Despite all efforts made to identify antigen targets that are suitable for antibody-based therapies in cancer, very few have been found that meet those criteria. Nevertheless, safe and effective ADCs have been developed, as indicated by the approval of brentuximab vedotin (Adcetris) in 2011 and trastuzumab emtansine (Kadcyla) in 2013. There is, however, considerable scope for better ADCs to be developed.
The Antibody
The antibody binding a tumor cell-surface antigen must activate receptor-mediated endocytosis for an ADC to enter a cell. Selection of internalizing antibodies is not always straightforward because different antibodies against the same target are not necessarily all taken up under the same conditions (1). Once inside a cell, an ADC has to be delivered to the lysosomal compartment for drug release. That occurs through degradation of the linker or antibody, so the intracellular trafficking of an ADC is an important determinant of its efficacy. Once the drug is released, it binds to its molecular target, usually DNA or tubulin, to effect cell death (2).
Immunogenicity: Therapeutic antibodies, whether they be chimeric, humanized, or fully human, contain amino acid sequences that are not in the human germ line, making these antibodies potentially immunogenic. Development of antitherapeutic antibodies (ATAs) has been well described (3). In some instances ATAs can bind to the variable domain of a therapeutic antibody, preventing it from binding to its target and therefore neutralizing its efficacy. The complexity of an ADC raises additional risks of an ATA response against the antibody, linker, or drug that can potentially lessen efficacy through reduced half-life, neutralization, or preventing effective internalization of the ADC. The risk of immunogenicity can be reduced by rational therapeutic antibody sequence design — in particular, avoidance (or removal) of CD4+ T cell epitopes that are essential for development of an adaptive humoral ATA response in patients (4).
Preclinical immunogenicity assessment and antibody engineering technologies can help select lead therapeutic proteins with the lowest risk of immunogenicity and/or avoid presence of CD4+ T cell epitopes in the variable domain of therapeutic antibodies, respectively. Such technologies have been pioneered at Antitope (a subsidiary of Abzena). Its Composite Human Antibodies technology creates therapeutic antibodies that do not incorporate CD4+ T cell epitopes in the variable domain but retain the functionality of the starting antibody. Selection of a lead antibody and confirmation of its lack of immunogenicity can then be performed using Antitope’s ex vivo T cell assays (EpiScreen), which have previously been shown to correlate with the development of ATA responses observed in clinical-stage products (5, 6). Such technologies provide a useful immunogenicity risk-mitigation strategy for ADCs, which pose an increased risk of an immunogenic response in patients from the addition of drug and linker to the therapeutic antibody.
The Drugs
The precursors of ADCs were recombinant antibody fusions with immunotoxins, such as Pseudomonas exotoxin (PE) and ricin-A. When the first ADCs were produced, the targeting antibodies were chemically conjugated to small-molecule cytotoxic drugs such as methotrexate and doxorubicin. Those first ADCs were not sufficiently potent at the intracellular concentrations that could be achieved with linker technologies available at the time.
More potent cytotoxic drugs such as the maytansinoids and auristatins, which have IC50 values in the pico-to-nanomolar range, are now typically used. They constitute the majority of ADCs in clinical development. However, because of their increased toxicity, it is critically important that they remain attached to a therapeutic antibody until it is internalized by a target cell and that the number of drug molecules attached to the ADC is carefully controlled.
Immunogenicity: Although recombinant antibody fusions with immunotoxins offered the benefit of high potency and low complexity during manufacture (requiring no chemical conjugation), early versions of antibody immunotoxin fusion proteins were highly immunogenic. Their use in treatment was often limited to a single dose before toxin activity was neutralized by antitoxin antibodies.
Deimmunization of immunotoxins (engineering to remove T- and sometime B-cell epitopes) may be a solution to reduce immunogenicity and enable development of this class of drugs as potential therapeutics. Deimmunization technologies (such as the Composite Proteins technology developed at Antitope) have been applied to produce immunotoxins with reduced preclinical immunogenicity and high activity. Such products include diphtheria toxin (Angelica Therapeutics), bouganin (Viventia), and PE38 (Ira Pastan, NIH). The progression of one or more of these immunotoxins into large-scale clinical trials should provide a resurgence of interest in using immunotoxins to treat cancer.
The Linkers
Early ADCs, such as gemtuzumab ozogamicin (Mylotarg) used acid-labile hydrozone linkers that are cleaved in the acidic environment of the cellular endosome rather than through systemic blood circulation (pH 5 vs 7.4) (7). The product is no longer marketed, but in 2000, it was the first ADC to receive regulatory approval in the United States. Disulfide-based linkers have also been used and are selectively cleaved in the cytosol by the more reductive intracellular environment (8).
Currently, peptide linkers such as the dipeptide citrulline-valine, which are selectively cleaved by lysosomal proteases, are increasingly being used and are proving to be more stable in serum than hydrozone linkers (7).
MMAE (monomethyl auristatin) is released from brentuximab when the valine-citrulline linker is enzymatically degraded in the lysosome by peptidases such as cathepsin B.
Noncleavable linkers also can be used for some toxin payloads. For example, DM1 (maytansinoid derivative) is released from trastuzumab (Kadycla) when the antibody is proteolytically degraded after internalization in the cell. The development of more selectively degradable, serum-stable linkers is one of the most important improvements made during the evolution of ADCs. Depending on the conjugation chemistry used, however, the drug can still be cleaved from the ADC before it reaches the tumor cell.
Conjugation
The most frequently used approach to conjugating drugs to therapeutic antibodies is by attachment to either lysine residues or thiol side chains of cysteines (obtained by reducing interchain disulfide bonds). Conjugation to lysine residues can be achieved by reaction with reactive groups such as a succinimidyl moiety to form a stable amine bond. For the Kadcyla product, this method is used to conjugate DM1 to trastuzumab (8). However, because a therapeutic antibody may have more than 80 solvent-accessible lysine residues available for drug conjugation, this method often produces a highly heterogeneous mixture of antibodies with different numbers of drug molecules attached (drug-to-antibody ratio, or DAR).
By contrast, there are fewer cysteine residues in an antibody, thus fewer sites for conjugation. An intact IgG1 antibody has four accessible interchain disulfide bonds that can be reduced to release eight free cysteine thiols for conjugation. Reduction of the interchain disulfide bonds is a critical step in the process because variations in reduction efficiency will yield two, four, six, or eight thiols that can be targets for drug conjugation in any particular antibody. Even though thiol-specific conjugation offers improvements in homogeneity compared with lysine conjugation, heterogeneous ADCs with DAR values ranging between 0 and 8 are still produced. Some such species are highly undesirable, particularly when antibodies are present with no drug attached (DAR = 0). Those will competitively inhibit binding of antibodies conjugated to the drug to the target antigen. High-DAR (>4)
ADCs are also undesirable because they have been shown to be less well tolerated and have higher plasma clearance rates and decreased efficacy in vivo (9). Various ways of controlling the sites of conjugation and reducing the number of drug molecules attached to an antibody have been investigated, with varying levels of success. Those include reengineering the antibody to include a single cysteine residue as the site of conjugation (10).
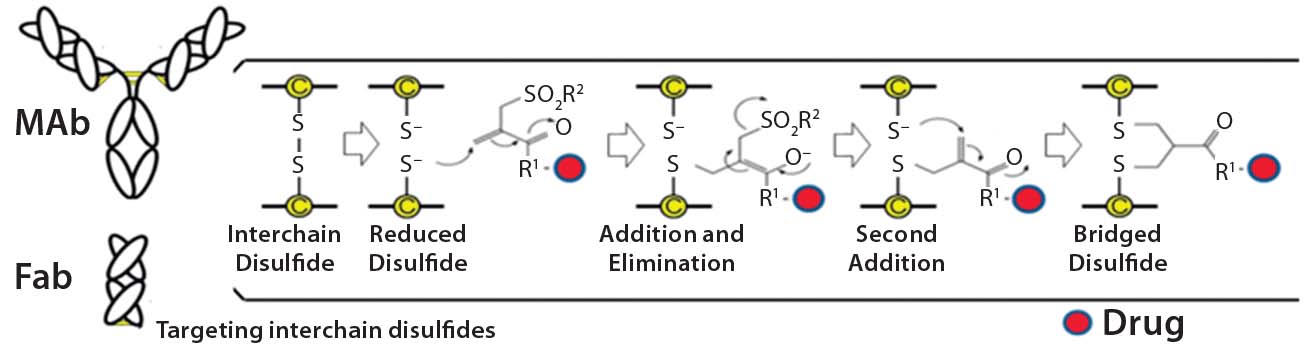
Figure 1: Conjugation of a drug to a disulfide bridge using a bis-alkylating conjugation approach
involving a sequence of Michael addition and elimination reactions; the disulfide is rebridged.
Heterogeneity: PolyTherics (a subsidiary of Abzena) has developed a novel way to prepare more homogeneous ADCs. The approach uses bis-sulfone reagents that are selective for the cysteine sulfur atoms from a native disulfide bond (Figure 1). The antibody interchain disulfides are selectively reduced, and the reagent undergoes bis-alkylation to conjugate both thiols derived from the two cysteine residues of the reduced disulfides. The reaction results in covalent rebridging of the disulfide bond by means of a three carbon bridge, leaving the protein structurally intact (11). The drug is attached to the bis-alkylating linker through a polyethylene glycol (PEG) chain to produce the reagent (ThioBridge technology) for conjugation. The reagent can undergo reaction at each of the four reduced interchain disulfides of an IgG1 antibody to produce ADCs that are predominantly DAR4, with minimal amounts of unconjugated antibody and DAR > 4 (Figure 2). Generation of predominantly DAR4 ADCs using this technology has been demonstrated with a range of different antibodies, using different drugs and with cleavable and noncleavable linkers (Figure 2).
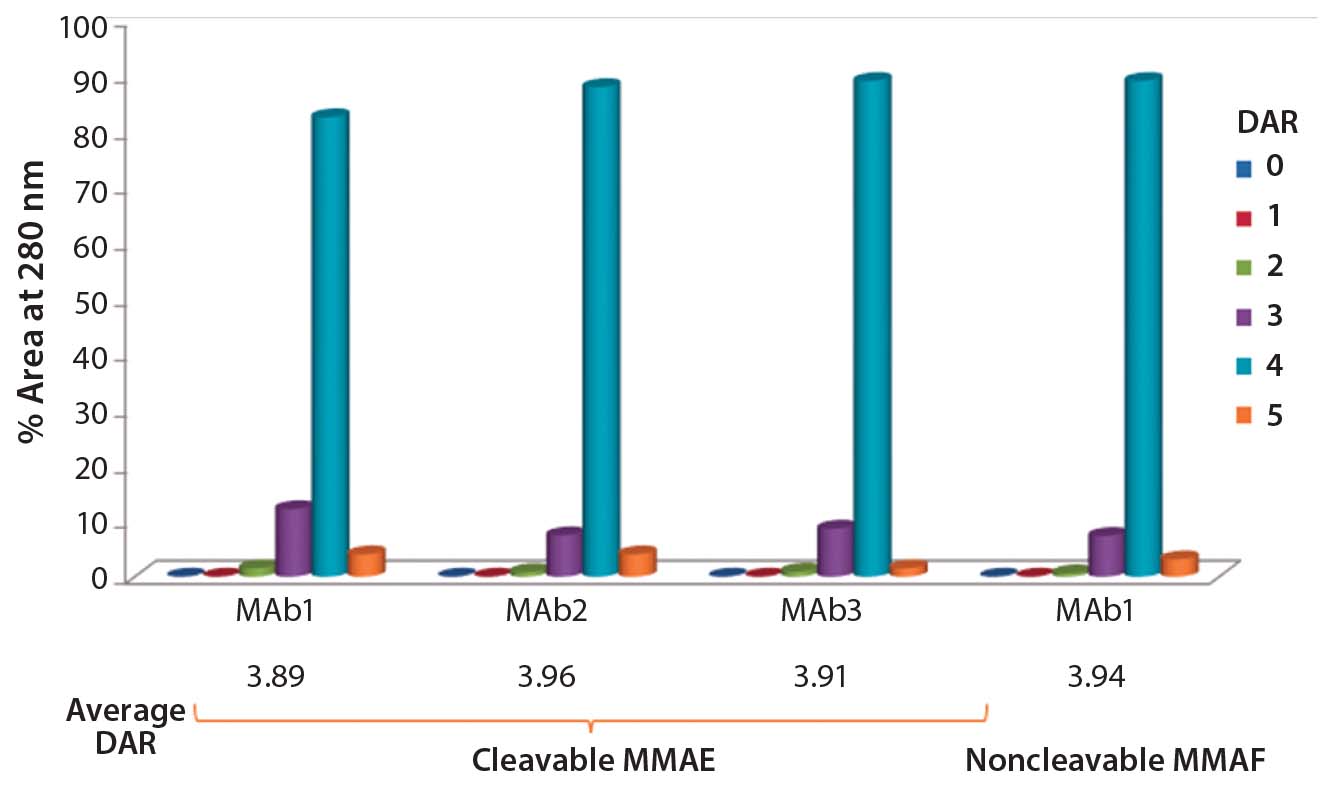
Figure 2: DAR distribution of ADCs produced by conjugation of MMAE to three different
antibodies using ThioBridge reagent with a valline-citruline (vc) linker and of MMAF to one of these antibodies using a noncleavable linker; analyzed using hydrophobic interaction chromatography (HIC); DAR4 was the predominant species.
Instability: ADCs produced using maleimide reagents are reportably unstable in serum due to deconjugation of the drug from the antibody and its subsequent cross-conjugation to the free-thiol groups of circulating albumin (10). Drug can also be released from an ADC if the antibody degrades before it reaches the tumor. As disulfides are reannealed with the ThioBridge conjugation reagent, the resulting ADCs should be more stable than maleimide conjugates.
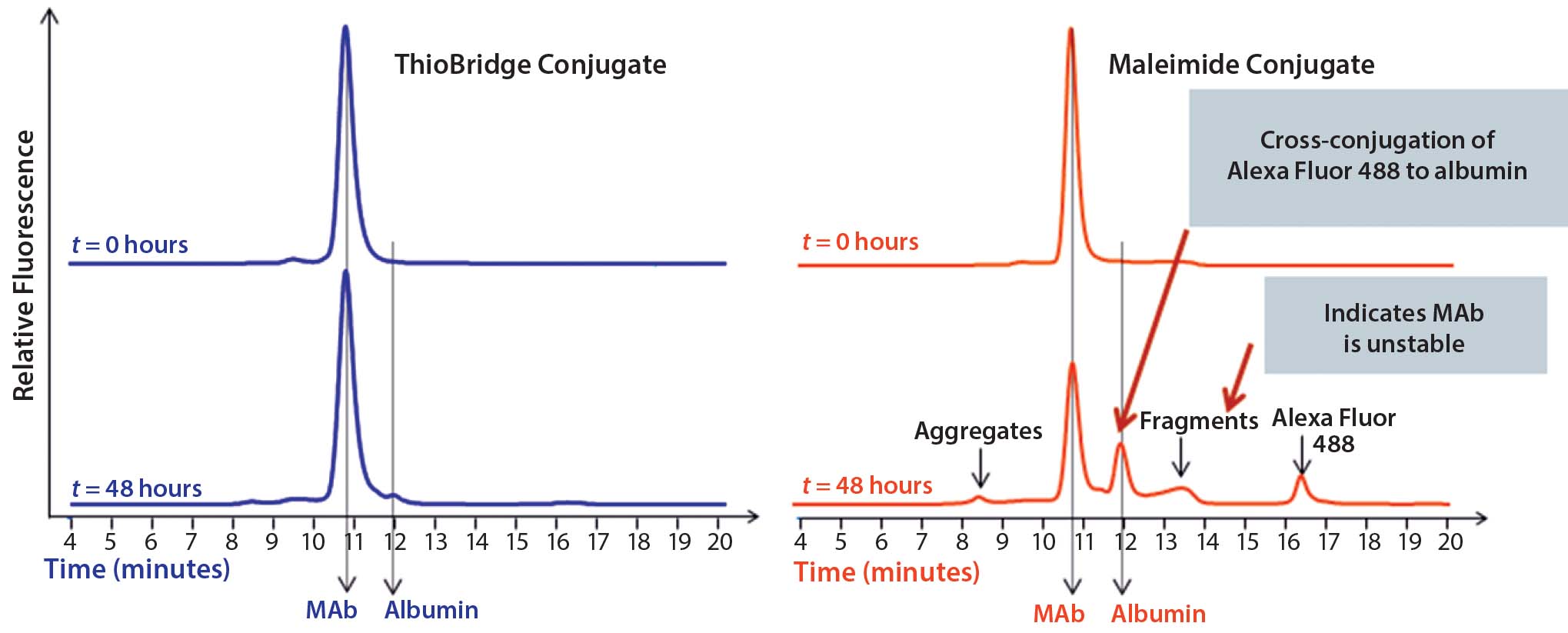
Figure 3: Comparison of the stability of ADCs produced by conjugating Alexa Fluor 488 to trastuzumab using either ThioBridge or maleimide reagents and incubated in rat serum at 37 ⁰C for 48 h; analyzed using size exclusion chromatography; the ThioBridge ADC was stable.
In stability studies in rat serum, the dye Alexa Fluor 488 was lost from the ADC when it was conjugated to trastuzumab using a maleimide reagent. By contrast, an equivalent ADC produced using a ThioBridge reagent was stable with no detectable crossconjugation of Alexa Fluor 488 to serum albumin (Figure 3). In addition, degradation of the antibody in the maleimide ADC was evident, but no degradation was seen in the ThioBridge ADC. IgG-depleted human serum had a clear difference in stability between maleimide and ThioBridge ADCs. The former showed a reduction in the proportion of DAR4 ADCs and concurrent detection of ADCs with DAR1, 2 and 3, and unconjugated antibody (Figure 4).
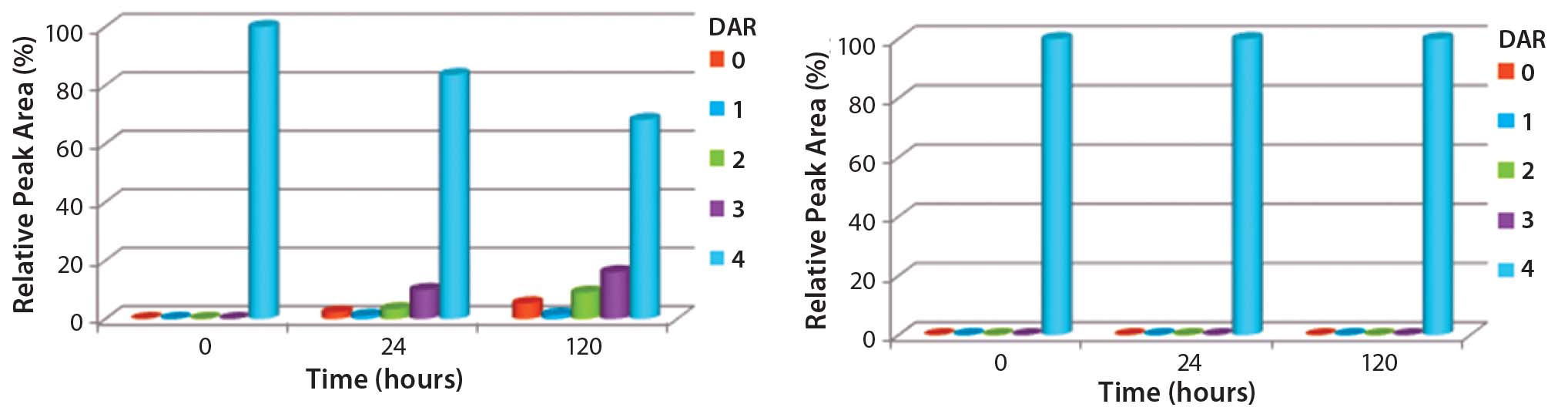
Figure 4: Comparison of the stability of ADCs produced by conjugating MMAE to trastuzumab using either ThioBridge or maleimide reagents and
incubated in IgG-depleted serum at 120 h for 37 ⁰C; HIC was used to determine changes in DAR distribution. Drug was lost from the maleimide ADC but not the ThioBridge ADC.
Next-Generation ADCs
The development of more homogeneous ADCs with better linker and antibody stability and reduced risk of generating an immune response should be the future goal to improve patient treatments. Most ADCs in development are intended to treat hematological cancers. One key reason for that is the poor penetration of full-length antibodies into solid tumors, which would limit the efficacy of ADCs made from such antibodies. The use of different types of targeting proteins to maximize tumor penetration is an area being explored to improve treatment of solid tumors.
The Thiobridge technology profiled here is applicable to any protein with accessible reducible disulfides and thus can be used to conjugate drugs to a range of different antibody formats and other tumor-targeting proteins. The method is particularly useful for conjugating drugs or imaging agents to antibody fragments (Fab), because a Fab has only a single interchain disulfide. This means that a highly pure and homogenous (DAR1) conjugate can be produced in essentially quantitative yield. PolyTherics also has conjugated drugs to other novel antibody formats and to tumor-targeting proteins for its collaborators.
The next generation of better ADCs still may be years from reaching the market. But with the improvements that have already been made and the new technologies now becoming available, cancer patients should have many more treatment options in the future.
References
1 Firer MA. Antibody–Drug Conjugates in Cancer Therapy — Filling in the Potholes That Lie Ahead (editorial). OA Cancer. 1(1) 2013: 8.
2 Trail PA. Antibody–Drug Conjugates As Cancer Therapeutics. Antibodies. 2(1) 2013: 113– 129.
3 Wolbink GJ, et al. Dealing with Immunogenicity of Biologicals: Assessment and Clinical Relevance. Curr. Opin. Rheumatol. 21(3) 2009: 211–215.
4 Baker MP, et al. Immunogenicity of Protein Therapeutics: The Key Causes, Consequences and Challenges. Self/Nonself 1(4) 2010: 314–322.
5 Baker MP and Jones TD. Identification and Removal of Immunogenicity in Therapeutic Proteins. Curr. Opin. Drug Discovery Dev. 10(2) 2007: 219–227.
6 Barker S. Immunogenicity of Biotherapeutics: A Need for Consensus on Flexibility of Approach (guest editorial). Current Drug Safety. 5(4) 2010: 272–274.
7 Beck A, et al. The Next Generation of Antibody–Drug Conjugates Comes of Age. Discovery Medicine 10(53) 2010: 329–390.
8 Phillips L, et al. Targeting HER2- Positive Breast Cancer with Trastuzumab-DM1, an Antibody–Cytotoxic Drug Conjugate. Cancer Res. 68(22) 2008: 9280–9290.
9 Hamblett KJ, et al. Effects of Drug Loading on the Antitumor Activity of a Monoclonal Antibody–Drug Conjugate. Clin. Cancer Res. 10(20) 2004: 7063–7070.
10 Shen BQ, et al. Conjugation Site Modulates the In Vivo Stability and Therapeutic Activity of Antibody–Drug Conjugates. Nat. Biotechnol. 30(2) 2013: 184–189.
11 Badescu G, et al. Bridging Disulfides for Stable and Defined Antibody–Drug Conjugates. Bioconjug. Chem. 25(6) 2014: 1124– 1136. c
Dr. Sally Waterman is senior vice president, corporate development, at Abzena plc, Babraham Research Centre, Babraham, Cambridge CB22 3AT UK; sally.waterman@ abzena.com; www.abzena.com.