The cell therapy industry continues to make progress, as measured by increasing numbers of clinical trials and patients treated (1). Although discussions of the differences between “off-the-shelf” (allogeneic) and “patient-specific” (autologous and matched allogeneic) therapies continue, we are confident that both will find success.
The best way to approach development for a cell therapy product is to consider these three fundamental drivers that guide development:
-
Speed to market (Which pathway will allow for fastest access to the commercial market?)
-
Operational efficiency (Which option provides the most efficient use of operational and financial resources?)
-
Reduction of risk (How can a company reduce the many risks involved in development of its new cell therapy?)
The clinical trial process is intended to provide a means by which candidate therapies can be proven to be safe and efficacious. Once a technology (e.g., chimeric antigen receptor-transduction, CAR-T) or cell type (e.g., mesenchymal stem cells, MSCs) has been targeted for development, a company wants to generate data as quickly as possible to support its safety and efficacy profiles. Figure 1 illustrates a typical development and commercialization pathway for a product candidate.

For cell therapies, product attributes heavily rely on manufacturing processes. So commercialization is unlikely to be successful without an effective development process. Companies use a number of approaches to optimize process and product development.
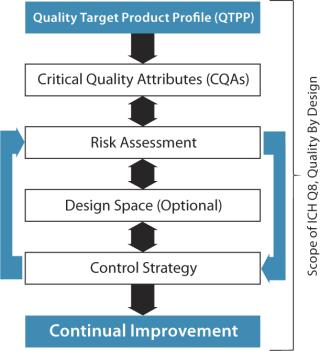
Figure 2 outlines components recognized by the US Food and Drug Administration (FDA) and other regulatory agencies regarding target product profiles and quality by design (QbD). Figure 3 goes a step further to define development by design (DbD), whereby critical aspects of quality, cost of goods sold (CoGS), scale, and sustainability are each addressed. Our company has formally adopted this DbD approach, building on its experience with cell therapy development over the past two decades.
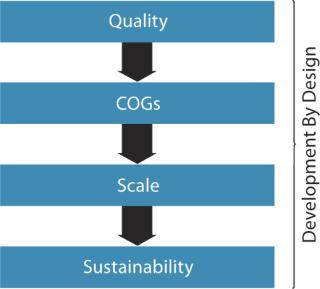
Attributes and Challenges of Development By Design
Quality is foundational, as recognized by QbD. And for cell therapies (which rely heavily on their manufacturing processes to meet final-product critical quality attributes) the manual, open, and human-dependent nature of many process steps presents substantial risk. A manufacturing process is only as strong as its weakest link. Thus, in the example of a patient-specific product, the strength of the process is directly related to reducing the risk of failure to treat the patient. Automation, integration, and closed-system designs are key tactics to elevate process robustness.
CoGS: The current high CoGS for cell-therapy products — typically driven by labor and testing costs for patient-specific products and media for off-the-shelf products — demands a sizable commercial value proposition. As processes mature, the focus on CoGS for commercial viability becomes critical. DbD allows for prospective approaches to address CoGS as appropriate for a given scale and stage of development.
Scale: Migrating from a clinical-scale process capable of making tens to hundreds of patient doses per year to a commercial-scale process with the capacity to make thousands to tens of thousands of patient doses can present significant comparability risk. In particular, cell-therapy products inherently possess high complexity, with one or more mechanisms of action that are often incompletely understood. In addition, there is currently a lack of analytical tools or in-vivo models for judging product comparability.
Sustainability: Even when quality, CoGs, and scale objectives are met, there can be a very real risk that manufacturing cannot be sustained over a full product life cycle. For example, a key risk is disruption of the relatively fragile and immature supply chain currently supporting the cell therapy industry. A disruption could halt manufacturing for an extended period. In the worst-case scenario, one process step relying on supply chain elements that become unavailable could require changes to be developed, tested, and comparability demonstrated. To mitigate risks to business sustainability, companies need to assess the full range of supply chain inputs to their manufacturing processes: reagents, consumables, equipment, and human resources. Additionally, such assessments should include every unit operation methodically, both process and testing.
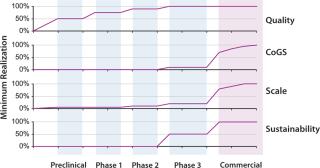
A key challenge in all development programs is that, although quality realization must occur early in development and be well-established by phase 2 trials, realization of the other aspects listed above is not required until much closer to commercialization (Figure 4). This puts significant pressure on cell-therapy developers to defer their investments in CoGS, scale, and business sustainability — until, in some sense, it may be too late. Developers often defer such investment until the comparability risks of making changes to address related concerns become substantial. Additionally, extreme changes in process scale becoming necessary as a developer moves toward commercialization can quickly create quality risks that were not yet encountered.
DbD is intended to guide strategy, provide structure and discipline to plan for, and address commercialization risks while there's still time to address them. A successful DbD program is implemented in the context of three drivers: speed to market, operational efficiency, and reduction of risk. It can be done in a way that is appropriate to each development stage. The goal should be to meet an ideal commercial manufacturing vision of providing consistently high product quality at reasonable cost that meets demand over the commercial life of the product.
Contract Manufacturing Option
Once a company has licensed or discovered a technology or cell type, that becomes a primary asset for the organization. Many companies choose to maintain their research internally so they can develop the best understanding of their technologies or cell types. Such knowledge is considered to be essential for development, troubleshooting, and fundamental scientific understanding. However, contracting for process development and manufacturing expertise may be a preferred option that can effectively support the drivers discussed above.
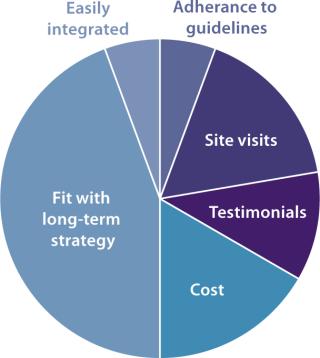
A recent industry report from Pharma IQ described the most important factors that developers consider when choosing a contract development and manufacturing organization (CDMO) (Figure 5). Interestingly, cost was not the most common reason for choosing one contract manufacturer over another. The report showed that “fit with long-term strategy” was far more important in making the choice. The same report identified the following as top concerns when working with a contract manufacturer: communication, teamwork between client and CMO, compliance with all emerging regulations, an ability to work within restrictive time frames, compliance with due diligence, adequate technology and experience, and an ability to protect intellectual property.
Case Study
The following case study shows how speed to market, operational efficiency, and risk reduction can be achieved by contracting out process development and manufacturing to a CMO/CDMO.
A private company was operating in virtual mode on technology that was spun out from an academic research laboratory. The company was able to produce enough material using that academic laboratory to conduct a small phase 1 study that helped it secure funding to support the company launch in anticipation of a phase 2 clinical program. The next clinical trial would treat 36 patients over the course of six months. A production model had been developed in which the company calculated that a minimum number of people would be required for both product manufacturing and facility maintenance. And management set a goal of six lots per month.
The company evaluated two options: to set up in-house manufacturing and stop operating in virtual mode or to outsource manufacturing (to PCT Cell Therapy Services) and continue virtual operations. The development program was divided into four phases (Table 1):
-
Project set-up, defining facilities, quality control (QC), staffing, and project needs
-
Technology transfer in which training, documentation, and materials sourcing are defined
-
Implementation and process qualification (PQ) involving facilities and process-specific qualification, definition of analytical methods and manufacturing processes, and qualifying shipping procedures
-
GMP manufacturing and production for the specific program.
Table 1
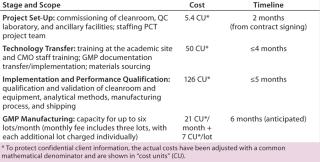
If we compare costs and duration for implementing that program at PCT and in-house to prepare for phase 2 manufacturing, we begin to see differences between the two options.
The primary benefits come during the first two steps: project setup and technology transfer. Those are routine operations for a contract manufacturer that has gathered related expertise by executing a number of other projects. For small/virtual companies, however, such efforts can present major challenges because they need to create facilities and processes, hire and train staff, and establish routine procedures. Larger, more-established companies may already have such experience in-house upon which they can capitalize. Table 2 compares costs and timeline efficiencies for this case study with an experienced contract manufacturer against performing the same services in-house.
Table 2
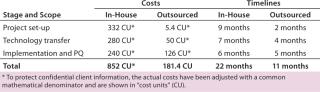
Aside from benefits realized during program implementation, engaging a CMO can be an efficient way to mitigate the resource strain that arises from unpredictable clinical trial patient accruals. The cost of maintaining “idle” capacity remains one of the most significant and common cost drivers related to cell-therapy manufacturing. The deeper and more sophisticated is the capacity, the higher will be its cost burden to the program. Expertise and capacity at a CMO can be used on a pay-per-use or minimum-retainer basis to reflect changes in accrual levels; in-house manufacturing must be maintained at a constant baseline even during low-accrual periods. Tables 3 and 4 illustrate how, in several accrual scenarios, the flexibility of engaging a CMO can dramatically reduce the costs of manufacturing resources and infrastructure.
Table 3
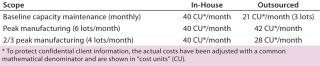
Table 4

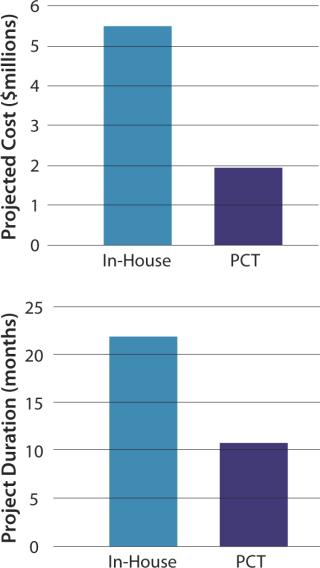
Thus, cost and timeline efficiencies for this case study are significantly greater when using an experienced contract manufacturer than when performing services in-house (Figure 6).
A Path to Success
As the cell therapy industry grows and matures, many companies seek the most efficient model by which to develop their products. Speed to market, operational efficiency, and reduction of risk are three main drivers for business model choices. If companies choose to outsource development work to a CMO, “strategic fit” will be a main factor in how they make their selection among the pool of available partners. Our case study represents how one sample company benefited specifically in speed to market and operational efficiency by outsourcing their development and manufacturing.
Author Details
Robert Shaw is vice president of commercial development; Brian Hampson is vice president of engineering and innovation; and Sanjin Zvonic, PhD, is director of technology services at PCT, 4 Pearl Court, Suite C, Allendale, NJ 07401; 1-201-883-5300;
REFERENCES