Commercial-scale viral vaccine manufacturing requires production of large quantities of virus as an antigenic source. To deliver those quantities, a number of systems are used for viral replication based on mammalian, avian, or insect cells. To overcome the inherent limitations in production outputs with serial propagation of cells, mammalian cells can be immortalized, which increases the number of times they can divide in culture. Modifications that immortalize cells are typically accomplished through mechanisms similar to those converting normal cells to cancer cells. Thus, the presence of residual host-cell nucleic acids in final vaccine products would create significant concerns about the potential for transfer and integration into a patient’s genetic material.
PRODUCT FOCUS: VACCINES
PROCESS FOCUS: DOWNSTREAM PROCESSING
WHO SHOULD READ: PROCESS ENGINEERS, QA/QC, ANALYTICAL
KEYWORDS: CHROMATOGRAPHY, BIOCATALYSIS, TANGENTIAL-FLOW FILTRATION, NUCLEIC ACID DETECTION ASSAYS
LEVEL: INTERMEDIATE
Host-cell nucleic acids in feed material depends on cell/virus type and on methods and techniques used in harvesting. The presence of DNA can contribute to process fluid viscosity and fouling of separation media, reduce useable capacity, cause coprecipitation, and threaten product safety. The risk of oncogenicity and infectivity of host-cell nucleic acid can be minimized by suppressing its biological activity. That can be achieved by decreasing the amount of residual DNA and RNA and reducing their size (with enzymatic/nuclease or chemical treatment) to below the functional gene length of ~100 base pairs.
DNA Removal
Precipitation with
-
Cationic detergents — e.g., cetyltrimethyl ammonium bromide (CTAB) or domiphen bromide (DB)
-
Short-chain fatty acids (e.g., caprylic acid)
-
Charged polymers — e.g., polyethyleneimine (PEI) and polyacrylic acid (PAA)
-
Polyethylene glycol (PEG)
-
Ammonium sulfate
-
Tri(n-butyl)phosphate (TNBP) with Triton X-100 detergent solution
Filtration:
-
Normal-flow filtration (NFF) with depth-charged or diatomaceous-earth–containing media
-
Tangential-flow filtration (TFF)
-
Ultrafiltration/diafiltration (UF/DF)
Chromatography and membrane adsorbers:
-
Anion-exchange chromatography (AEX)
-
Gel-filtration (size-exclusion) chromatography
-
Hydrophobic charge-induction chromatography (HCIC)
Degradation with
-
Enzymes
-
Physical forces (shearing)
-
Alkylating agents (e.g., β-propylactone)
Health authorities and regulatory bodies such as the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) have set limits for acceptable amounts of residual DNA in final biological products. According to requirements published by the FDA, a parentally administered dose is limited to 100 pg of residual DNA. The EMA and the World Health Organization (WHO) allow 10 ng per parenteral dose and 100 μg/dose for an orally administered vaccine (1). Orally administered DNA is taken up about 10,000× less efficiently than parenterally administered nucleic acid.
The biopharmaceutical industry continues to improve its purification processes to minimize potential risks associated with harmful immunological and biological responses caused by residual impurities originating from host cells and culture media. Here we describe a new method for removal of host-cell DNA and/or RNA impurities that offers some advantages over known approaches. We also summarize our development of a process incorporating this new approach.
A typical cell-culture–based viral vaccine production process begins with propagation of a selected “seed” cell line. When the culture has grown to a predetermined cell density, the virus of interest is introduced (inoculated) and begins to replicate. A few days later, virus is harvested directly from the host cells, from the culture supernatant, or both.
When virus is harvested from the host cells directly, a cell disruption step is required to release intracellular viruses. Available methods for cell disruption fall into two main categories: chemical (detergents and surfactants, enzymes, osmotic shock) and physicomechanical (heat, shear, agitation, sonication, freeze-thawing). Mammalian cells are easy to break, so the methods used for their disruption are relatively mild and may include treatments like use of detergents and/or hypotonic buffer.
When virus is harvested from supernatant, there is no need for a cell breakage step; the supernatant can be directly clarified from cellular debris. If a virus must be harvested from both cells and supernatant, collected cells will be disrupted before the viral suspension is clarified.
Methods for Nucleic Acid RemovalThe amount of host-cell–related impurities (including nucleic acids) in a process fluid varies significantly depending on the methods used for cell lysis and/or virus harvest. The “DNA Removal” box lists several techniques that can be applied for reduction and/or removal of genomic DNA from cell culture process streams. But all of those methods are limited in the types of products and processes for which they could be applied.
Purification of viruses from cell substrate components such as DNA is a particularly challenging task in viral vaccine production for a number of reasons:
-
Physical similarities of viruses and nucleic acids could limit the resolution and selectivity of an applied method. For example, similar electrical charge (both viruses and nucleic acids are typically negatively charged at neutral pH) and size create limitations in separation with chromatography and filtration methods (Table 1). And sedimentation behavior similarities could lead to coprecipitation.
-
Applied techniques and reagents could affect biological virus activity and integrity, causing losses of infectivity and/or potency due to degradation by physical forces (shear) or chemicals (detergents). Caprylic acid, for example, can inactivate enveloped viruses.
-
Some methods can cause nucleic acids to bind with a product of interest (virus/glycoprotein/protein), which lessens the efficiency of downstream purification processes in achieving the necessary level of final-product purity and compromising process yields and economics.

Most of those limitations and challenges can be minimized when the amount of nucleic acid contaminant is reduced using enzymatic degradation with endonucleases. Such enzymes act by specifically catalyzing the hydrolysis of the internal phosphodiester bonds in DNA and RNA chains, breaking the nucleic acids into smaller nucleotides. Naturally present in bacteria, these enzymes defend cells from invasion by foreign DNA. The several types of these nucleases are derived from different sources.
A New ApproachSerratia marcescens is a Gram-ne
gative pathogenic bacterium that secretes (among other proteins) a very active endonuclease that cleaves all forms of DNA and RNA (single-stranded, double-stranded, linear, and circular) without sequence specificity. The nuclease cleaves nucleic acids very rapidly, with a catalytic rate almost 15× faster than that of deoxyribonuclease I (DNase I) (2). The enzyme shows long-term stability at room temperature and is active in the presence of both ionic and nonionic detergents as well as many reducing and chaotropic agents. But it has proteolytic activity of its own. All these characteristics make this enzyme useful for biotechnological and pharmaceutical applications.
A genetically engineered form of Serratia nuclease (Benzonase from Merck Millipore of Darmstadt, Germany) is made using Escherichia coli and recombinant technology. The Benzonase enzyme is a dimer of identical subunits with molecular weight ~30 kDa each (with a weight totaling ~60 kDa). Its isoelectric point is at pH 6.85, and it is functional in a pH range of 6–10 and at temperatures of 0–42 °C. The presence of Mg2+ (at 1–2 mM concentration) is required for this enzyme’s activity.
The Benzonase enzyme digests all forms of nucleic acid by hydrolyzing them into smaller oligonucleotides of <10 base pairs in length (1 bp is ~650–660 Da) with minimal sequence specificity. (Hydrolysis occurs slightly more often in guanosine and cytosine (GC)–rich areas than in adenine and thymine (AT)–rich sequences). During a typical reaction, nucleic acid molecular weights are reduced as in Figure 1 through ethidium-bromide–stained agarose electrophoresis gels.
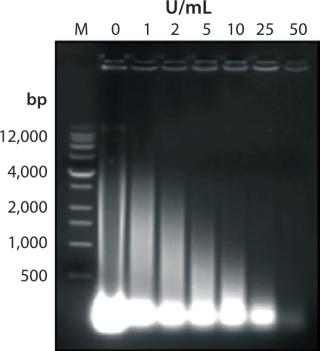
One Benzonase unit is defined as the amount of enzyme that causes a change in UV absorbance at 260 nm of one absorption unit within 30 minutes. That unit degrades about 37 μg DNA in 30 minutes.
Process DevelopmentBecause process times for enzymatic reactions often measure in hours, Benzonase treatment is typically carried out in batch mode. To start a reaction, the enzyme is added to a process feed. Large amounts are often used to ensure maximal digestion of nucleic acid impurities. In such cases, no optimization of the enzymatic reaction is needed; process step optimization takes into account temperature, exposure time, and shear on product stability rather than on specific Benzonase activity.
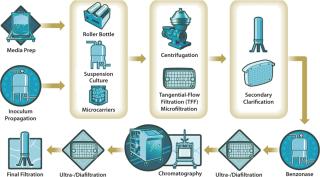
Current examples for laboratory scale applications demonstrate use of 9–90 U/mL. If required, the enzymatic reaction can be optimized through studying reaction rates in microwell plates because only very small quantities are needed both for reaction and DNA/RNA detection. After treatment, subsequent purification steps must quantitatively remove the enzyme from a process stream. Benzonase treatment therefore should be placed sufficiently upstream in the overall process (Figure 2).
Critical parameters of the enzymatic reaction to consider during process development include enzymatic activity, DNA and RNA feed and target concentrations, process time, enzyme and Mg2+ concentrations, temperature, pH, and the presence (and concentration) of Benzonase inhibitors and multivalent or monovalent salts in media and their concentrations. And typical enzymatic reactions can be described by Michaelis– Menten kinetics (
The volumetric rate of reaction (vrr) is proportional depending on the maximum reaction rate at infinite reactant concentration (vrrmax) and on the substrate concentration (S) of nucleic acids DNA and RNA and the Michaelis constant (Km).
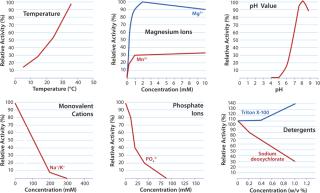
The highest Km is achieved by operating at optimal pH (8–9) and temperature (37 °C). Although higher temperatures expedite the reaction kinetics, the need for simplified control and/or concerns over product stability often lead to operating at room temperature or lower. Mg2+ ions are needed to get optimal enzymatic activity.
Another critical parameter is the enzyme’s starting activity, so enzyme concentration is provided in units (as described above) rather than actual concentrations. When concentration of host-cell nucleic acids in starting feed material is high, the maximum reaction rate is independent of the their concentration. As enzymatic digestion progresses and nucleic acid concentration is reduced, the reaction will become first order and the removal rate is reduced.
Some process additives and agents affect Benzonase activity. The enzyme can be inhibited by high salt concentrations (Figure 3): >300 mM monovalent cations, >100 mM phosphate, >100 mM ammonium sulfate, or >100 mM guanidine HCl. Other known inhibitors include chelating agents. EDTA, for example, could cause loss of free Mg2+ ions (EDTA concentrations >1 mM have shown to inhibit the enzymatic reaction), an effect that can be reversed by adding more MgCl2. And the presence of components such as 4M urea could have an opposite effect and increase Benzonase activity.
Equation 1
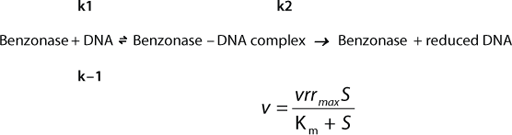
Because the starting material might contain specific inhibitors, concentrations, or salt choices — and enzyme quality (in terms of activity) is predefined — process optimization at a given position in the process can focus on controllable variables as a function of process time: enzyme and Mg2+ concentration, temperature, pH, and salt co
ncentrations. The information herein can be used for troubleshooting purposes for cases in which excessive amounts of enzyme are used.
Regulatory authorities do not regulate how much residual endonuclease can be in a vaccine product. However, vaccine manufacturers using it in their processes need data to demonstrate safety/toxicity status and measure residual endonuclease that might be present in final preparations. Consider Merck & Company’s EU patent of VAQTA hepatitis A vaccine (3). It indicates that residual Benzonase enzyme is lower than 0.0001 ng/dose. It is important to note that the endonuclease is a process additive and not a drug, excipient, or active pharmaceutical ingredient.
Benzonase removal from a vaccine process stream can be accomplished by several downstream unit operations, so Benzonase treatment is often positioned in the “upstream” part of processing. Removal can be demonstrated by showing a lack of residual nuclease activity (which does not detect residual nonactive enzyme) and using an enzyme-linked immunosorbent assay (ELISA) for detection of total residual Benzonase molecules (both active and nonactive).
Irreversible Benzonase inactivation occurs within ~15 min at a temperature >70 °C and 0.02 N NaOH. Such conditions could negatively affect the integrity of viral vaccines, however. Heating the product solution also could increase Benzonase activity, thus affecting the determination of residual nucleic acids in samples. Removal of the enzyme can be accomplished using classical downstream methods, as described below. A clearance technique can be chosen to align with additional purification steps for the vaccine.
Tangential-Flow Filtration (TFF): Benzonase enzyme is removed in filtrate while viral particles are retained. For this ~60-kDa molecule, we suggest 300-kDa Biomax or larger cut-off sizes. Depending on the viral particle size (viruses should be retained with low passage), TFF might be a possible option. An internal study at our company indicated that Benzonase enzyme could be removed by diafiltration ≤99.5% at five diavolumes and >99.9% after eight diavolumes using 300-kD membrane. The overall diafiltration profile is close to a theoretical sieving value of 1. We recommend 300 kD as an acceptable molecular-weight cut-off (MWCO) value for the Benzonase clearance after DNA digestion in viral cell cultures — provided that the membrane is retentive enough for the virus of interest.
Case Study
Size/amount estimation of Benzonase spike required for treatment of cell substrate for gDNA digestion for a live/attenuated virus for injectable vaccine product
Virus propagation system: adherent VERO cell culture
Cell lysis type: chemically induced
Product stream: postsecondary clarification (NFF) viral suspension
Host cell/gDNA content: ~1 μg/mL
Batch volume (product stream of interest): ~10 L
Total amount of gDNA: 10,000 μg
Minimum Benzonase amount for gDNA digestion: ~270 U (0.027 U/mL)
Benzonase type to apply: HC, >99% pure, 25 KU, Catalog#71206-3 (250 U/μL)
Benzonase amount/spike applied: 500 U (=2 uL)
Typical amount used: 20 U/mL (minimum) to 50 U/mL (maximum)
Safety factor used: 740 (minimum) to 1,852 (maximum)
Anion-Exchange Chromatography (AEX) is considered to be the most convenient and effective chromatography technique for Benzonase removal. With a pI of 6.85, the enzyme will typically flow through while a viral product is bound to an AEX column or (if it is bound as well) elutes separately. Table 2 lists several applicable AEX resins using a number of sample and equilibration buffers.
Table 2: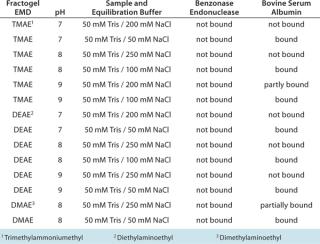
Cation-Exchange Chromatography (CEX) has been shown to remove Benzonase enzyme, but the operating range might be smaller than for AEX. Table 3 lists a few CEX chromatography media and conditions that are suitable for the Benzonase removal.
Table 3: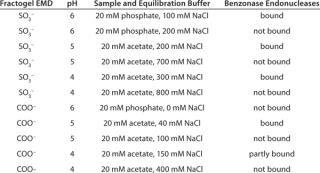
As chemical composition, process time, and process temperature are critical parameters, it is important during scale-up to ensure that the entire solution is mixed efficiently so that all parts of a batch experience similar enzymatic reaction conditions. That can be achieved using well-characterized mixers and thermal control. Understanding mixing and temperature equilibration is critical in reaction scale-up. Take care to ensure that treated material is not contaminated with untreated material (e.g., residual material in a transfer line or splashed material that is not part of the batch reaction). Some viruses are shear sensitive, so confirmation of viral product stability during mixing needs to be verified. Mixing speed thus might be explored as an area of focus. The “Case Study” box above provides a typical example of Benzonase use sizing.
DNA and RNA AssaysThe residual DNA limit of 100 pg/dose set by regulatory authorities equals the DNA amount from ~17 diploid Chinese hamster ovary (CHO) cells (4). Detection of such a small amount of DNA requires an extremely sensitive and robust analytical method. The European Pharmacopoeia advises that residual DNA should be determined using sequence-independent techniques (5). Common methods of measuring DNA include UV absorbance at 260 nm (maximal absorbance with an extinction coefficient of 50), fluorometric detection using ethidium bromide (EtBr) or Hoechst 33258, a Picogreen assay (Life Technologies), quantitative polymerase chain reaction (qPCR), and a Threshold DNA assay (Molecular Devices). Table 4 summarizes their detection limits.
Table 4: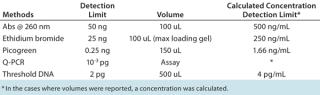
Components in the feed mixture may affect assay results. To assess the level of digested DNA bands, ultralow-range DNA ladders — ≥10 bp according to polyacrylamide gel electrophoresis (PAGE) or agarose-electrophoresis measurements — could be used (6)(7). Before qPCR analysis is performed, Benzonase enzyme must be inactivated or removed; otherwise, it may digest newly amplified DNA. Adding an ice-cold solution of perchloric acid (4%) will instantly stop the Benzonase activity. The Threshold DNA assay is not based on hybridization, but rather relies on reaction chemistries and luminescence. Reported detection limits are as low as 4 pg/mL (2 pg for a 0.5-mL sample). DNA log reduction factors (LogRF) can be calculated using
Equation 2
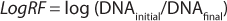
Several methods are used for removing nucleic acids from bioprocess streams, all of which are still valid for current processes. But new cell lines pose challenges to drug safety. It is important to eliminate host cell DNA/RNA impurities by thoroughly removing them from a product stream. Benzonase enzyme offers some advantages over competing methods of nucleic acid removal. Not designed to be part of the final product, however, it needs to be removed after use.
Benzonase removal from a vaccine process stream can be accomplished using several types of downstream unit operations. Depth filtration for clarification, TFF for concentration and diafiltration, and chromatography for purification can also remove nucleic acids. The latter two could be used to ensure Benzonase removal from treated product streams. Results can be demonstrated by a lack of residual nuclease activity and with an ELISA for detecting total residual endonuclease.
Author Details
Elina Gousseinov, MS, is a process development scientist at EMD Millipore in Canada. Willem Kools, PhD, is head of field marketing and biomanufacturing sciences at EMD Millipore in Bedford, MA. And corresponding author Priyabrata Pattnaik, PhD, is director of the worldwide vaccine initiative at Merck Millipore, 1 Science Park Road, #02-10/11 The Capricorn, Singapore 117528; 65-6403-5308;
Priyabrata Pattnaik, PhD
ARTICLE IS VERY USEFUL TO KNOW SERIOUS NESS OF RESIDUAL BIOLOGICAL IMPURITY
THANKS FOR VERY GOOD ARTICLE PUBLISHED
pl check my article
Quantification and Quantification Residual biological …
http://www.oatext.com/pdf/VRR-1-127.pdf · PDF file
Correspondence to:Kailas Narale, Sr Manager Quality Assurance, , India, Tel: +919860363666; E-mail: kvnarale@gmail.com Received: September 18, 2017; Accepted: October 23, 2017; Published: October 25, 2017 When implementation by regulators Quantification and Quantification Residual
Dear authors,
Thank you for this wonderful work. I have a question regarding the removal Benzonase. It was claimed that “residual Benzonase enzyme is lower than 0.0001 ng/dose” . Could you please provide its reference? I am interested in how was such low concentration determined and it would be greatly helpful for us. Thank you!
Dongsheng