The recent success of monoclonal antibodies (MAbs) as therapeutic agents to treat cancer, multiple sclerosis, rheumatoid arthritis, and other chronic inflammatory and autoimmune disorders (Table 1) has catapulted these once difficult-to-develop molecules to the forefront of modern molecular medicine (1, 2). The size of the global MAb market in 2008 was valued at almost US$28 billion. Industry analysts predict that the size of the MAb market will grow to almost $68 billion by 2015, with the largest growth occurring in oncology and chronic inflammatory diseases (2).
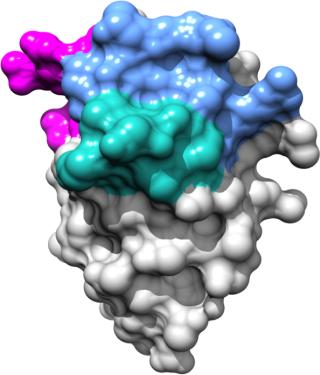
Table 1: Commercialized monoclonal antibodies in the United States and European Union
The first MAbs to be commercialized in the early 2000s were chimeric MAbs (genetically engineered hybrid molecules containing murine and human sequences) and “humanized” MAbs (containing 90–95% human antibody protein sequences). Since their introduction, many of those products — including Remicade, Enbrel, and other drugs — have attained blockbuster status.
PRODUCT FOCUS: MONOCLONAL ANTIBODIES, FRAGMENTS, AND OTHER PROTEINS
PROCESS FOCUS: ANTIBODY ENGINEERING
WHO SHOULD READ: PRODUCT DEVELOPMENT, ANALYTICAL
KEYWORDS: ANTIBODY FRAGMENT ANALYSIS; FLOW CYTOMETRY, IMMUNOHISTOCHEMISTRY; RADIOLABELING; PROTEIN FUSION
LEVEL: INTERMEDIATE
The commercial success of those early products prompted huge investments by pharmaceutical and biotechnology companies in therapeutic MAb drug development. Such investments — coupled with major advances in MAb engineering technology — led to development of fully-human MAbs (molecules that contain only human antibody protein sequences).
Fully human MAbs generally exhibit improved therapeutic efficacy and possess fewer side effects (better safety profiles) than chimeric or humanized MAbs. To that point, Humira (Abbott) — one of the first fully human MAbs to be marketed as a treatment for rheumatoid arthritis and other chronic inflammatory indications (Table 1) — is one of the world’s top-selling drugs with sale projected to exceed $9.3 billion in 2012 alone (3). Currently, ~70 chimeric and humanized MAbs and 30 fully human MAbs are in various stages of clinical development worldwide (2).
Some Clinical Limitations of Therapeutic MAbsAlthough MAbs are one of the biotechnology industry’s greatest commercial successes to date, they possess several characteristics that can limit their utility and clinical effectiveness (4, 5). First, their complex molecular structure (four polypeptide chains — two light and two heavy), glycosylation of heavy chains, and at least one disulfide bond in each of several immunoglobulin domains require that full-size antibodies be manufactured in eukaryotic or mammalian expression systems (e.g., CHO cells). That process can be extremely labor intensive and costly (6). Second, their large size (150–160 kDa) makes them unsuitable for most intracellular therapeutic targets, so MAbs must be delivered to patients by injection or infusion (7). Third, most MAbs (because of their large size) are limited in their ability to penetrate and accumulate in tissues. That can reduce their effectiveness in treating some types of cancers (e.g, solid tumors). In some cases, MAbs are unable to bind to epitopes on the surface of protein targets that are accessible only by smaller-sized molecules (8). Finally, the complex molecular structure of MAbs typically renders them thermally unstable, which requires that they be stored at refrigeration temperatures to ensure stability and clinical efficacy (5).
Because of such limitations, scientists over the past 10–15 years began to develop novel and alternative MAb engineering platforms. Such platforms can be divided into two major groups: antibody-derived and nonantibody-derived protein scaffolds. Both have yielded smaller molecules that possess most of the desirable features of MAbs (e.g., antigen specificity and binding) but lack many of the limitations mentioned above. Some advantages that protein scaffolds offer over MAbs include small size (12–50 kDa), high thermal stability and solubility, better tissue penetration and accumulation, low production or synthesis costs, and intellectual property protection and freedom to operate around high value MAb-directed targets (7,8,9).
Antibody-Derived Protein ScaffoldsSmall antibody-based formats derived from preexisting MAbs were the first type of protein scaffolds to be developed. These include single-domain (dAbs), nanobody (from camelids), single-chain variable fragment (scFv), antigen-binding fragment (Fab) (10), Avibody, minibody, CH2D domain, Fcab (11, 12), and bispecific T-cell engager (BiTE) molecules. All those are thermally stable, soluble, and maintain their binding and antigenic specificity. They are less costly to produce than conventional MAbs because they can be chemically synthesized or manufactured using nonmammalian expression systems. Of all such molecules, most clinical progress has been made with scFvs and nanobodies (Table 2).
Table 2: Antibody-derived scaffolds
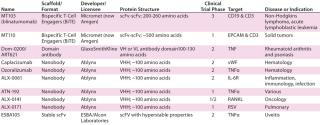
ScFvs are single-chain antibody fragments (28 kDa) that contain variable regions of MAb heavy (VH) and light (VL) Ig chains fused together (using antibody engineering) by a flexible peptide linker that usually contains 10–25 amino acids (10). Nanobodies (12–15 kDa) are antibody fragments derived from naturally occurring heavy-chain–only antibodies from camels and llamas (9, 13).
ScFvs have been used mainly in flow cytometry and immunohistochemistry applications (13). However, two BiTE-based scFv molecules are in clinical development for various oncology indications (Table 2). In contrast, six nanobodies in phase 1–2 clinical development are being developed as treatments for infectious diseases, a variety of cancers, and several chronic inflammatory diseases (Table 2) (9).
Nonantibody-Derived Protein ScaffoldsNonantibody-derived scaffolds use naturally occurring nonantibody binding proteins (Table 3) that can be genetically engineered to specifically recognize and bind to potential therapeutic target antigens (5, 8, 9, 14). The naturally rigid protein structure of these scaffolds allows their existing binding sites to be modified (or create a new site) to recognize and bind to prescribed antigenic targets (5).
Table 3: Types of non-Ig scaffolds

Over the past decade or so, more than 50 different nonantibody protein scaffolds have been proposed as alternative MAb development platforms (5). Of those, about 10 different protein classes (Table 3) are most advanced, including several in late-stage clinical development. They include
-
engineered Kunitz domains of human serine protease inhibitors (e.g., LAC1-D1), which can be genetically engineered for different target protease specificities (15)
-
DARPins (designed ankyrin repeat domains): small, engineered single-domain proteins that can be selected to bind to any protein target (16, 17)
-
avimers created from multimerized low-density lipoprotein receptor class A (LDLR-A) (18)
-
anticalins derived from lipocalins (which are abundant in humans, insects and many other organisms) that naturally form binding sites for small ligands by means of four structurally variable loops found in the proteins (19)
-
knottins constructed from cysteine-rich knottin peptides (11, 20)
-
affibodies that are based on the Z-domain of staphylococcal protein A (21)
-
adnectins or monobodies and pronectins based on the 10th or 14th extracellular domain of human fibronectin III, which can adopt an Ig-like β-sandwich fold with two or three exposed loops without a central disulfide bridge (14, 22).
Other nonantibody protein scaffolds of interest include Fynomers and nanofitins. Fynomers are 7-kDa binding proteins derived from SH3 domains of the human Fyn tyrosine kinase. Along with bispecific Fynomers, they are being developed by Swiss-based biotechnology company Covagen AG (23). Nanofitins (formerly Affitins) are structurally derived from the DNA binding protein Sac7d found in Sulfolobus acidocaldarius, an archeabacterium (24). In addition, affilins derived from one of two proteins — gamma-β crystallin (a family of 20-kDa proteins found in the eye lenses of vertebrates, including humans) or ubiquitin (a highly conserved 76-kDa protein found in eukaryotes) — are being used as a non-Ig scaffold by German company Scil Proteins, GmbH to develop new biopharmaceutical drugs (25).
Commercialization of Protein ScaffoldsNonantibody-derived scaffolds can in principle be used to create highly specific antibody-like molecules that recognize and avidly bind to therapeutic targets. However, only one product, Kalbitor escallantide (or DX-88), has been successfully commercialized to date (26).
Escallantide (manufactured by Dyax Corporation) was approved in 2009 by the US Food and Drug Administration for treatment of hereditary angioedema (HAE) and prevention of blood loss in cardiothoracic surgery (Table 4) (27, 28). It is a 60–amino acid polypeptide engineered from a Kunitz domain that inhibits the enzyme kallikrein (26). Kallikrein is a plasma-based protease that mediates the release of bradykinin from its protein precursor kininogen (26). Excess bradykinin leads to fluid leakage from blood vessels, causing swelling of tissues characteristic of HAE. Ecallantide inhibits the pathogenic mechanism by selectively and reversibly inhibiting the activity of plasma kallikrein (5, 29).
Table 4: Non-Ig scaffolds in development

Progress has been made to commercialize other nonantibody scaffolds, including DARPins and anticalins (Table 4). MP0112, a DARPin directed against vascular endothelial growth factor (VEGR) cellular receptors, is being developed by Switzerland-based Molecular Partners as a possible treatment for various ophthalmologic disease indications (17). The molec
ule was evaluated in phase 1 clinical trials and is currently entering phase 2 testing. Likewise, Pieris, a Germany biotechnology company, has developed an anti-VEGR anticalin designated PRS-050 (Angiocel) as a potential treatment for certain solid tumors. Angiocel has been evaluated in phase 1 clinical trials.
Other nonantibody scaffolds, including avimers and affibodies, have not fared as well as DARPins and anticalins. After acquiring Avidia in 2006, Amgen sought to advance the company’s lead product candidate AMG-220 (C326), an avimer that neutralizes the proinflammatory cytokine interleukin-6 as a possible treatment for Crohn’s disease. Although AMG-220 successfully completed phase 1 testing, Amgen subsequently halted clinical development of the molecule (5, 30).
One major disadvantage of affibodies as potential therapeutic agents is that they are derived from protein A, a bacterial protein that generates an immune response after repeated administration to patients. Although affibodies will not lend themselves to development as therapeutic agents, attempts have been made to develop them as molecular imaging agents for cancer detection. For example, a radiolabeled affibody molecule Z (HER2:342) was successfully used to image HER-2 positive tumors in murine models to better identify and select cancer patients who might benefit from Herceptin (trastuzumab) treatment (31,32,33,34).
Monobodies derived from the 10th or 14th extracellular domains of human fibronectin III (FnIII) are perhaps the most clinically advanced nonantibody protein scaffolds in development today (Table 4) (8, 14, 22, 35,36,37,38). Adnexus (a subsidiary of Bristol Myers-Squibb) is evaluating Adnectin CT-322 (BMS-844203), a VEGFR-2 inhibitor, as a treatment for glioblastoma, pancreatic cancer, and other oncology indications (36).
Results from phase 1 clinical studies showed that CT-322 was safely administered at 2 mg/kg and exhibited promising antitumor activity without severe toxicity in patients with advanced solid tumors (37, 38). The molecule is currently in phase 2 clinical testing (in combination with irinotecan, 5-flurouracil, and leucovorin). It is being compared with Avastin (bevacizumab) as a secondary treatment for patients with metastatic colorectal cancer (39).
Protelica (a small Northern California-based biotechnology company) has created a new FnIII-based scaffold using the 14th extracellular domain of human fibronectin III. This unique scaffold has been used to develop a series of proprietary molecules called Pronectins, which are being evaluated as possible treatments for various clinical indications.
The most advanced Pronectin molecules — several of which have been evaluated in preclinical toxicology and ADME studies — are directed against various cellular antigens, including AXL (metastatic cancer), VEGF-R2 (macular degeneration, various cancers), and Frizzled receptors (stem cell differentiation and cancer) (40). Protelica has taken three of its lead candidate molecules through preclinical development and hopes to advance them into clinical testing within the next few years.
Limitations of Protein ScaffoldsOne major disadvantage of smaller protein scaffolds is their rapid renal clearance and correspondingly short circulating serum half-lives. Typically, proteins smaller than 50–60 kDa have serum half-lives of less than one hour (12). Many approaches have been used to increase the serum half-lives of various protein scaffolds by increasing their molecular size. Strategies include fusion to albumin or antibody fragments, adding a second scaffold that binds to albumin, attaching half-life extension peptides, and PEGylation (17, 41, 42).
Domantis (now GlaxoSmithKline) developed a technology called serum albumin binding domain antibodies (AlbudAbs) that was successfully used to increase the serum half-life of the interleukin-1 receptor antagonist IL-1a (following fusion to an AlbudAb) from minutes to about four hours in mice (43). Likewise, an IL-6-specific avimer fused with a domain that binds to IgG molecules exhibited an extended serum half-life (18). PEGylation of Adnectin CT-322 was used to improve its pharmacokinetics and overall clinical utility (36). Similarly, after PEGylation, the Kunitz-type inhibitor DX-1000 (which inhibits plasminogen activation) showed improved pharmacokinetics and tumor modulating properties (28).
Another disadvantage of protein scaffolds is their lack of effector functions typically provided by MAb Fc regions (e.g., complement-dependent cytotoxicity, antibody-dependent cellular cytotoxicity, (ADCC), and antibody-dependent cellular phagocytosis (ADCP) (44). In some cases, effector functionality may be required to provide a therapeutic benefit to molecules engineered using protein scaffolds. To that end, Fc fragments or other effector molecules to protein scaffold can easily be fused using conventional antibody engineering technologies. That method is a facile solution to add effector functions if needed (44).
All protein-based therapeutics scaffolds are potentially immunogenic. Consequently they may elicit unfavorable immune responses that could either neutralize or interfere with intended therapeutic actions of drugs. To obviate this possibility, most protein scaffolds are derived from naturally occurring human proteins. Atlhough that does not eliminate the possibility of unfavorable immune responses, it minimizes the likelihood that the effectiveness of therapeutic agents derived from protein scaffolds will be dampened by neutralizing or other immune antibodies.
Promising AdvancementsThe importance of MAbs as treatments for cancer, chronic inflammatory diseases, and autoimmune indications is growing every year. Although therapeutic MAbs represent viable treatment options for many diseases, they are not without their shortcomings as therapeutic agents. That has prompted researchers to develop a variety of “next-generation” antibody engineering platforms, including antibody- and nonantibody-derived protein scaffolds and antibody–drug conjugate (ADC) technologies.
Although protein scaffolds and ADCs are still in their early stages as therapeutic agents, they offer unique opportunities to create novel drugs to combat difficult-to-treat diseases with rapidly growing unmet medical needs. The utility of these molecules as therapeutic agents will evolve as they continue to move their way through clinical development and ultimately comercialization.
Author Details
Corresponding author Clifford S. Mintz is president and founder of BioInsights, Inc.;
REFERENCES
rcepharma.com/special-reports/humira-0 9.