Vaccines are among the most efficacious and cost-effective human health interventions available. They provide protection against a surprisingly broad spectrum of infectious diseases. Notable recent successes protect against human papillomavirus (Cervarix and Gardasil vaccines from GlaxoSmithKline and Merck, respectively) and rotavirus (Rotarix and RotaTeq vaccines from GlaxoSmithKline and Merck, respectively). However, generating reliable sterilizing or therapeutic immunity is still not possible against a number of latent and chronic pathogens that especially affect people in developing countries. Among those agents are Mycobacterium tuberculosis, human immunodeficiency virus, hepatitis C virus, and the Plasmodium protists that cause malaria.
Protection against such complex pathogens may require activation of cellular immunity, a compartment of the human immune system that most conventional vaccines usually fail to target. An exception to that rule are live attenuated vaccines, but the approach is unavailable or unsuitable for many global communicable diseases. In addition, for certain attenuated viruses, there is a risk of reversion to pathogenic strains (1) and potential residual virulence for some vaccine recipients or contact persons of them (2, 3). Such risks are unacceptable in the modern era of vaccines, for which increases in both international mobility and numbers of immunocompromised individuals demand a greater degree of safety in the vectors (4,5,6).
PRODUCT FOCUS: VACCINES
PROCESS FOCUS: MANUFACTURING
WHO SHOULD READ: QA/QC, PROCESS DEVELOPMENT, AND MANUFACTURING
KEYWORDS: CELL CULTURE, FED BATCH, CELL LINE DEVELOPMENT, FILTRATION, DOWNSTREAM PROCESSING, SCALE-UP
LEVEL: INTERMEDIATE
Modern vectored vaccines use carriers unrelated to their target pathogens to elicit a protective immune response against recombinant antigens (7). These combine the advantages of live vaccines with the strong safety profile of highly attenuated vectors and thus may provide novel therapeutic or protective approaches. Promising viral carriers include host-restricted pox viruses such as modified vaccinia Ankara (MVA) that trigger a strong immune response without an ability to replicate in humans. That is an important biological characteristic for safe application in immunocompromised recipients and heterogeneous populations.
But this type of attenuated vector cannot amplify at the site of inoculation, so relatively high numbers of infectious units per dose must be given to maximize efficacy (although much lower numbers than are required with adenovirus vectors). To maintain large stockpiles for emergency applications—or if such vectors are to be used in global vaccine programs—highly efficient production processes are required. Such processes must also be scalable, robust, and preferably autonomous. That is, production should be independent of complex logistics for supply of the cell substrate (as with egg-based technologies) and culture media.
Although some viral vectors have been used in clinical trials, currently no viral-vectored vaccine is licensed for use in humans, although many are licensed for veterinary use. Here, we describe development of MVA85A as a novel viral-vectored vaccine against tuberculosis (TB), focusing on solving production challenges to enable large volumes of vaccine to be produced at an affordable cost of goods (CoGs).
MVA85A and Tuberculosis
MVA is a host-restricted poxvirus shown to be well tolerated in a number of clinical trials (8,9,10,11,12). With very few exceptions (13,14,15), replication of MVA is restricted to avian cells. However, mammalian cells are susceptible to infection; although the virus cannot complete a full infectious cycle, a strong immune response is induced against the vectored antigen (11, 16,17,18). Even immunocompromised patients can receive these vectors, opening the potential for therapeutic vaccines against certain cancers and chronic or latent infectious diseases. One such MVA-based vaccine is MVA85A, an advanced TB vaccine candidate originally developed at the University of Oxford (19) and subsequently licensed exclusively to the Oxford-Emergent Tuberculosis Consortium (OETC) and Emergent Biosolutions (www.emergentbiosolutions.com) for development and commercialization.
TB is a devastating mycobacterial disease that, according to World Health Organization estimates, latently infects a third of the global population and caused 1.2–1.5 million deaths in 2010. It is second only to human immunodeficiency virus (HIV) as a leading cause of death from infectious disease worldwide (20). Immunosuppression, HIV infection, and malnutrition are additional risk factors for TB, and it has an enormous socioeconomic impact, especially in developing countries. A resurgence of TB in industrialized countries includes increasing levels of multidrug-resistant (MDR) and extensively drug resistant (XDR) mycobacterial strains. Therapy for infection with the new strains is difficult, and treatment failure in HIV-negative XDR tuberculosis patients is in the range of 40–70%, even with the highest standards of supervised therapy (21). Of great concern is the recent emergence of strains sometimes referred to as extremely drug resistant (XXDR-TB) or totally drug resistant (TDR) variants that respond to no conventional or second-line antibiotics (22). That development appears to have accelerated because patients with unrecognized MDR or XDR strains far too often receive inappropriate medication (23), further limiting their treatment options.
Epidemiologic modeling shows that reduction and elimination of TB cannot be achieved solely with existing vaccine and drug therapies. New, more effective, safe, and affordable vaccines are required (24). The current Bacille Calmette Guérin (BCG) vaccine is an live attenuated bovine tuberculosis species (Mycobacterium bovis) that protects against mycobacterial infection of the meninges. However, because BCG vaccination provides a variable protective benefit against the more common pulmonary form of tuberculosis (25), new vaccines are in development. BCG is currently a core component of the WHO’s Expanded Programme on Immunisation (EPI, www.who.int/immunization_delivery/en) and one of the world’s most widely used vaccines, with >100 million doses administered every year, primarily to infants.
MVA85A displays the mycobacterial component 85A to boost the human cellular immune response already elicited by BCG. Antigen 85A is an ideal vaccine target for several reasons. It constitutes a major portion of the secreted proteins of M. tuberculosis (26). It is highly conserved amongst all mycobacterial species and is a major target of BCG-induced immune responses (27, 28). And it has proven to be an essential gene in the final stages of mycobacterial cell wall assembly (28). MVA85A is being studied for use in BCG-vaccinated infants, adolescents, and young adults as well as in HIV-infected adults. This clinically advanced TB vaccine candidate has shown favorable efficacy data in four animal models and is well tolerated and immunogenic in more than
15 ongoing or completed clinical trials in adults, adolescents, infants, HIV-positive adults, and latently infected individuals. The first efficacy data for MVA85A in an infant population is expected in the fourth quarter of 2012.
Hyperattenuation of the MVA-based vaccines comes at a cost of the dose requirement, however: An estimated 108 infectious units per vaccination are required for efficient stimulation of the immune system (29, 30). For global programs against infectious diseases, hundreds of millions of doses of the highly attenuated poxviruses could be required annually. By contrast, lesser-attenuated strains with limited replication potential also produced on avian cells include vaccines against measles, mumps, and yellow fever; these require only 103 to 5.5 × 104 infectious units per dose (according to the package inserts of Sanofi Pasteur’s YF-VAX and Merck’s M-M-RII products). The protective dose of the Dryvax vaccinia strain in routine use against smallpox is 2.5 × 105 infectious units (31), which is 400-fold lower than that recommended for MVA-based vaccines. Hence, novel highly efficient and robust production systems for MVA-based vaccines (including MVA85A) will be required to reach all intended recipients.
Generation and Characterization of a New Host Cell Line
MVA and other vaccine strains adapted to avian hosts are still produced in embryonated hens’ eggs or on fibroblasts prepared from such eggs. This is an established technology, but it comes with numerous disadvantages (32). The chicken embryos have to be introduced continuously into the manufacturing process, which requires elaborate husbandry to keep donor flocks free of specific pathogens. Extremely complex logistics involving transport of material across country borders are required because the number of specialized animal facilities is limited.
Embryonated eggs are also required for production of live and killed vaccines against a number of indications (including seasonal and pandemic influenza, selected pediatric diseases, and rabies). New global vaccineprograms would therefore compete with established products for such demanding and expensive materials. Furthermore, seasonal differences and complexities in preparation could lead to lot variations (33, 34). Finally, even with several layers of precautions in place, contamination cannot always be prevented. Because time is short from collecting the embryonated eggs to vaccine production, testing for extraneous agents is performed on the final bulk product (35). Occasionally, whole vaccine lots are discarded when contamination is confirmed by quality testing (36). That places further strains on the supply of both vaccines and production substrates.
Those limitations make primary chicken cells a less than optimal choice for producing a modern and novel vaccine against a global infectious disease. For development of an MVA manufacturing process, ProBioGen established a new cell line by immortalizing primary cells from a single Muscovy duck embryo. Because of difficulties associated with purifying live viruses, the cell line was designed such that all products derived from it would have reduced risk of adventitious agent contamination.
The donor animal was a duck rather than a chicken because chickens are known to harbor endogenous retroviral particles that can copurify with a produced vaccine (37). Consequently, the company used no feeder cells to establish its primary culture, thus preventing introduction of rodent amphotropic viruses. For immortalization, a focused biochemical approach based on the early proteins of human adenovirus 5 was used (38), a ubiquitous virus that causes the common cold. Each step—from generation of the cell line to its adaptation to chemically defined media—is fully documented and performed in a dedicated clean room. And each substance that came into contact with the cells during those processes is linked to an appropriate quality certification.
Master and working cell banks (MCBs, WCBs) reserved for MVA85A manufacturing processes have recently been generated. They provide a level of consistency and reliability for continuous manufacture at industrial scales that cannot be achieved with chicken primary cell substrates. In accordance with regulatory guidelines, the absence of adventitous agents was confirmed before production through extensive testing of the cell banks with a spectrum of independent, overlapping, and complementary methods. Those include transmission electron microscopy (TEM) for detection of microbial structures; real-time polymerase chain reaction (PCR) against specific pathogens including circoviruses; cocultivation with sentinel cell lines for cytopathic effects caused by unknown pathogens; quantitative fluorescent product-enhanced reverse-transcriptase (QFPERT) assay and cocultivation against the presence of retroviral contamination; inoculation of cell line material onto agars and broths to test against mycoplasma and other bacteria; inoculation of chicks for antibody induction; and inoculation of embryonated eggs, guinea pigs, and adult and suckling mice for signs of infection.
Immortalizing duck rather than chicken primary cells provides a host that is free of endogenous retroviruses but presented a risk because virus permissivity (especially a highly attenuated virus such as MVA) cannot be inferred by phylogenetic relationship. Duck cells cannot be expected a priori to behave the same way as chicken cells, which are known to allow replication of that virus. Therefore, the duck cell line was tested early against MVA and other viruses to determine its permissivity (Figure 1). In those initial experiments performed on adherent cells (with serum present), MVA titers were already beyond the critical threshold of 108 infectious units per milliliter of culture. The clone of this Muscovy duck cell line chosen for further development was named AGE1.CR.pIX.
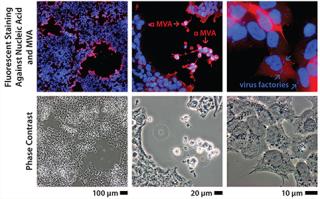
Upstream Process Development
At this phase in development we aimed to design a robust and highly efficient production process for host-restricted poxviruses. The process had to be freely scalable from intermediate to very large volumes in disposable bioreactors (≤2,000 L) and preferably independent of microcarriers and more elaborate processing steps such as perfusion. Media and buffers had to be free of animal-derived components or hydrolysates associated with regulatory hurdles or lot inconsistencies.
It is difficult to reconcile those requirements with an adherently growing cell line. The next step was to screen commercially available culture media for adaptation of AGE1.CR.pIX to cultivation in suspension. Cell proliferation was good in media from a number of suppliers, but infection of suspension cells with MVA resulted in burst sizes (the ratio of progeny to input virus) 39), which readily replicates in a range of cell proliferation media. But poxviruses use the cytoske
leton for spreading (40, 41), so spherical cells without polarized actin organization may inefficiently disseminate such viruses in culture. We tested virus replication using suspension media and adherent cultures (and vice versa). Burst rates were high for cell suspensions infected in medium for adherent cultures, but they were low in adherent cultures infected in suspension media. That result indicated a need to optimize the culture medium rather than the less accessible suspension host cell properties.
The next phase in developing an upstream manufacturing process was surprisingly complicated. The team used an extensive optimization matrix, but attempts to derive a medium that allowed both passaging of the avian cell lines and efficient MVA production failed. Virus production media allowed MVA replication but interfered with cell proliferation. Because a complete change of medium (perfusion) would be inconvenient in a commercial setting, the company targeted a fed-batch process with the proliferation medium supplemented by a virus production medium only at the time of infection. The final media are
- an optimized, chemically defined proliferation medium for the AGE1.CR.pIX cell line and
- a virus production medium (also chemically defined) for maximum production of virus at the time of infection.
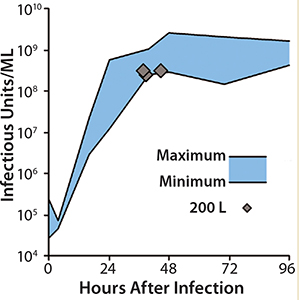
With that new method, yields of 109 infectious units/mL in the crude cell lysate were obtained within 48 hours after infection in 1-L and 10-L laboratory-scale bioreactors. Typical parameters such as pH and impeller frequencies were refined, and the potential for scalability was demonstrated in a 50-L disposable bioreactor. The next phase focused on technology transfer to industrial volumes.
Scale-Up to Industrial Volumes: Morphogenesis of MVA is complex; a significant portion of the infectious virions are not released into the supernatant. For the design of a manufacturing process, this property is an important difference between influenza virus and MVA. In suspension cultures the effective production of MVA was increased by the controlled formation of cell aggregates that are induced upon addition of the virus production medium (Figure 2). Although this process was extremely reliable, robust, and without failures at the laboratory scale, a transfer to industrial volumes of 200 L and beyond was not without risk. For example, after revitalization of a single cryovial of cells in a conventional shake tube/shake flask incubator, additional steps for cell expansion had to be introduced using a disposable wave-style bag reactor to generate sufficient cell numbers to seed a 200-L scale disposable stirred tank bioreactor. Due consideration was also paid to agitation and gassing strategies on scale up. Agitation profiles were designed to ensure efficient mixing at both the 100-L volume when cells are expanded in the growth medium and at the 200-L volume when an equal volume of viral production medium is added. The need to maintain adequate mass transfer for oxygen had to be balanced against the desire to limit specific power input (W/m3) to maintain cell aggregates after infection. The fact that the viral titers remained almost constant throughout all these process modifications on scale-up demonstrates the robustness of the cell line for production of MVA-based vaccines (Figure 3).
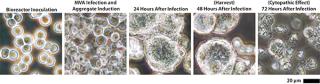
Downstream Process Development
Available purification techniques are limited for live viral vaccines, with few removal steps directed against adventitious agent contamination. So one considerable challenge is to perform the entire downstream process aseptically, using disposable technologies wherever possible. Additional challenges are posed by properties unique to poxviruses, including MVA. The viral particles are large, unwieldy flattened cylinders ~360 nm in their long axes (42). A considerable portion of mature and infectious virions are tightly cell associated (41, 43). Host cells remain largely intact even in late cytopathic stages, which further interferes with virus release.
Some MVA virus properties are used to an advantage in the downstream process by performing hollow-fiber tangential-flow filtration (TFF) for harvest
The next phase of processing begins with sonication to fully release viruses from the host cells. Normal-flow filtration (NFF) clarifies the sonicate to remove cell debris, proteins, and some residual cell DNA. The virus is further purified using a series of TFF steps and anion-exchange membrane absorption (to remove host-cell proteins and nucleic acids), as well as incubation with endonucleases to further degrade host-cell nucleic acid so it can be removed in further TFF steps. All buffers are optimized to prevent viral aggregation. At the end of this step, bulk material is once more clarified using TFF and added to the final buffer formulation. The resulting finished drug product is subject to stability and quality control (QC) testing.
Properties of the Final Preparation
A number of regulatory guidelines describe the requirements that vaccine preparations for human use should meet (44, 45). As briefly outlined above, extensive assays must be performed on the host cell line to thoroughly demonstrate the purity and identity of MCBs, WCBs, and end-of-production cell banks. Another set of guidelines describes properties of the master and working virus seed stocks and the final product. MVA85A identity is determined using PCR and Western blotting against the 85A antigen.
The fully chemically defined production process developed here obviates the need to screen for antibiotics or serum components that sometimes are added in primary cell cultures. With live vaccines, rescue of virus activity imposes restrictions on the stringency of the purification process, so reduction of host-cell–derived nucleic acids and proteins can be a significant technical hurdle. Current guidelines suggest fragmenting cellular DNA to obtain residual levels to ses and significant depletion of host-cell proteins.
The vaccine formulation is currently intended to be a lyophylate that is stable at 2–8 °C to facilitate transport and distribution.
On to Clinical Trials
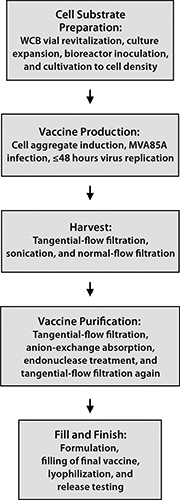
MVA85A is an advanced TB vaccine candidate in which a highly attenuated poxvirus serves as a carrier for expression of a mycobacterial antigen in recipients. One hurdle toward realization of this product for global health programs is making adequate numbers and concentrations of infectious units at an affordable CoGs. We successfully resolved this problem by designing a novel cell line that is permissive for the host-restricted poxvirus. The infectious cycle was transferred into a scalable suspension culture using chemically defined media, and a manufacturing process (Figure 4) was developed that yields virus at a concentration and purity suitable for clinical and potentially commercial application. Material generated using this process is designated for use in phase 1 studies with healthy adults followed by an age deescalation to infants and subsequent phase 3 infant studies to support licensure.
Author Details
Corresponding author Ingo Jordan and Volker Sandig are with ProBioGen AG, Goethestraβe 54, 13086 Berlin, Germany; 49-30-924-006-0, fax 49-30-924-006-19; ingo.jordan@probiogen.de; www.probiogen.de. Nigel Woods and Gary Whale are with Emergent BioSolutions, 540 Eskdale Road, Berkshire RG41 5TU, UK.
REFERENCES
<div
id=”CIT0032″>32.) Chua, JV, and WH. Chen. 2010. Bench-to-Bedside Review: Vaccine Protection Strategies During Pandemic Flu Outbreaks. Crit. Care. 14:218.