The cell therapy industry (CTI) is no longer a cottage industry; it is a distinct and sustainable component of the global healthcare sector (1). Today, CTI prospects are strong, with annual revenues exceeding US$1 billion/year, supported by improving investor sentiment and public support (1,–3). The next phase of CTI growth — toward a multibillion-dollar global industry — will depend on the biomanufacturing community innovating to meet growing market demands and providing products at affordable costs to healthcare payers.
Currently, the majority of cell therapy clinical trials are in phases 1 or 2 (4). However, as more CTI companies break the phase 3 frontier, clinical trials and regulatory requirements are becoming increasingly more predictable. This enables new companies to start out with a focus firmly on commercialization, using lessons learned from preceding trials to streamline their manufacturing processes and minimize the time to market.
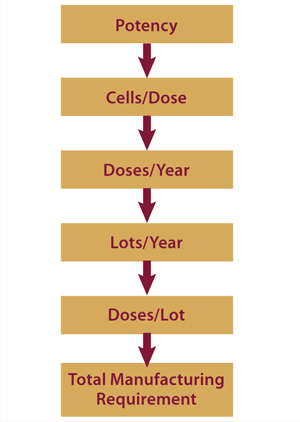
Here we propose a simple requirements-based framework for approaching three key, interrelated aspects of cell therapy bioprocessing that must be considered as these therapies translate from laboratory development to market launch: characterization, scale-up, and cost of goods (CoGs). In particular, we highlight the critical need to appreciate commercial implications of key bioprocessing decisions to maximize the efficiency and speed of cell therapy translation.
Characterization
Cell characterization is the process of identifying the relevant biological properties that define a population of cells. In cell therapy biomanufacturing, it is essential to understand the nature of the product being administered to patients and to ensure that it is the same every time. Essentially, this involves defining the biological aspects — the critical quality attributes (CQAs) — of a cell therapy product. Product characterization should begin in a laboratory setting before clinical trials so understanding can improve throughout clinical development and into commercialization.
All CQAs are crucial to thorough characterization, but they are discussed in detail elsewhere (9). However, potency is particularly relevant when determining manufacturing demand and will our focus here.
In development and preclinical studies, evidence typically emerges that indicates potential mechanism(s) of action (MoAs). For instance, if a therapeutic agent is antiinflammatory, then when it is injected into a mouse with an inflammatory condition, it should elicit improvements in the animal’s condition — indicating an antiinflammatory MoA. Evidence may even be generated that elucidates whether a product’s antiinflammatory properties are attributable to secreted factors (deduced using supernatant assays) or cell–cell contact (deduced using cell-based assays). Linking MoA to assays early on begins the process of product characterization with respect to potency. Several potential potency assays should be identified as early as possible and can subsequently be refined based on clinical data from phase 1 before qualification during phase 2 (5). That is crucial because the FDA stipulates that potency tests be established by phase 3 (6, 7).
In parallel with assay optimization throughout clinical trials, manufacturing processes undergo continuous development. However, considering that when deploying living cells as therapies — “the product is the process,” (8) — even minor tweaks most likely will give rise to changes in the final product. So the parallel activities of process development and assay qualification are complementary. By understanding which cellular parameters are essential to CQAs, it is possible to demonstrate product comparability alongside process development activities, which can potentially become qualified and validated concurrently (9).
It is best practice to have an arsenal of assays that test for characteristics beyond those strictly required for lot release. These additional assays — deemed “for information only” (FIO) — are not used for lot release, although they are indispensable as back-up assays if one desired lot release assay is not amenable to validation (e.g., the assay is too inherently variable) or if there are assay supply shortages (e.g., materials used to produce an assay are discontinued). Furthermore, FIO assays can be tracked throughout process changes to reinforce comparability or help identify noncrucial characteristics. For example, it is worth noting when a product stops producing a specific cytokine even if that does not affect its target function in patients.
Using preclinical studies to identify several tests that can be used to measure comparability throughout process development is a diligent strategy. It maximizes the chances of successful validation of potency tests before phase 3 clinical trials begin.
Commercial Implications: The central driver of early, thorough product characterization is to make the journey to commercialization as short as possible. Understanding a product not only enables competent scale-up, but it is also instrumental in defining release criteria. Many CTI companies, for example, require the services of a contract manufacturing organization (CMO) at some point. Product characterization will expedite the necessary technology transfer process later on. That ensures that CMOs can add maximum value by optimizing and scaling existing processes rather than providing essentially contract research services to address basic scientific deficiencies before they can proceed.
Potency tests are not only vital (required by the FDA for progression to phase 3), but they also have huge implications for a product’s commercial potential. Therapeutic potency correlates with the required number of cells per therapeutic dose. From predicted market size and capture, it is possible to estimate a required number of doses per year; from manufacturing capacity and processing time, it is possible to assess the number of lots per year. Those figures can be used to find required dosage per lot, which can then be used to design and implement a manufacturing strategy capable of achieving the necessary results (Figure 1).
For example, a therapy with a required 50 million cells/patient dosage and a treatment target of 100,000 patients (doses) per year might be manufactured at a rate of two lots/week (~100 lots/year). Each lot size would need to produce 1,000 doses (50 billion cells per lot) in addition to essential material required for quality control. Processing on this scale is feasible using certain current technologies such as larger culture tray units, hyperdense platforms,
and suspension cultures. However, for cases in which a larger dose is required — say 500 million cells — new technologies will be required that exceed the current gold standards by one or two orders of magnitude.
Regenerative Medicine Glossary
Cell therapy: Therapy in which cells are administered to the body to the benefitof the recipient
Whole bioprocessing: Allogeneic (entire bioprocess from donor through to implantation of a cell therapy) or autologous (entire bioprocess from patient biopsy through to implantation of a cell therapy)
Identity: To confirm that product contains the intended cellular and noncellular components
Potency: To confirm that the product possesses the inherent or induced biological function(s) that is relevant to treating the intended clinical indication
Purity: To confirm that the product does not contain undesired components, such as contaminating cell types or residual process reagents
Safety: To confirm that the product is not contaminated with microbes or adventitious agents and, if appropriate, does not have tumorigenic potential
References
1 PAS 84: Regenerative Medicine Glossary. Regen. Med. 44, 2008: S621–S622.
2 Carman J, et al. Developing Assays to Address Identity, Potency, Purity and Safety: Cell Characterization in Cell Therapy Process Development. Regen. Med. 2012: 85–100.
Scale-Up
Once a total manufacturing requirement has been established, a strategy to achieve it should be determined. There are different methods of scaling up, but as a general rule, because of cells’ inherent sensitivity to their environment, large-scale processes need to remain substantially similar to their original small-scale process for maintaining CQAs (10,–12). Thus, if cells grown for early clinical studies are cultured on planar surfaces (such as a T-flasks or trays), then on reaching larger-scale operations, those cells will need to be cultured similarly. But that may be difficult to implement and limit production capacity later on. Likewise, if a desired end point of a scaled-up process is to culture cells on microcarriers, then the product used in early studies ideally should have also been produced using suspension culture. If not, final product cells will probably not to be comparable to those used in clinical trials. This illustrates why product characterization is so important.
Many process changes are associated with cell culture, but do not underestimate the investment required — in skills, resources, and time — for downstream processing (DSP). It is an underappreciated fact that most cell therapy manufacturing processes involve greater bottlenecks in DSP than in cell culture (13). As these processes are scaled up, technological barriers in volume reduction and filling steps can significantly delay process completion. It is best to strategically plan the full-scale requirements of each unit process at each step on the road to commercial scale. Thus, large-scale DSP technologies for cell therapy bioprocessing can be developed and deployed at appropriate times, preventing the kinds of DSP bottlenecks that plague the protein industry today.
Process scale-up can occur at any point during clinical development. However, the regulatory burden incurred when applying such changes increases as a product progresses through the clinical pathway. In early stage trials, for example, the FDA may request comparability studies based on product characterization data; in later stages, clinical bridging studies may be stipulated. Therefore, it is prudent to optimize and lock down large-scale manufacturing processes early and certainly before commencement of phase 3 trials. That should decrease the number of regulatory and financial hurdles.
Even historically predictable scale-up strategies (e.g., from small to larger Nunc Cell Factory) can impart some unwanted cellular changes, such as the loss or gain of a certain marker or the halted or activated secretion of a particular cytokine. However, as mentioned above for characterization, it is sometimes the case that such changes resulting from scaling methodologies are irrelevant to a product’s clinical function. If it can be proved that given changes do not affect the proposed MOA or cause adverse side effects, then the new process is likely to be accepted by regulators. Agencies recognize this, which is why products need to be demonstrated as “comparable” and not “identical.”
Before locking down a process, consider several key widely applicable aspects (Table 1) to minimize contamination risks and ensure batch-to-batch reproducibility.
Table 1. Streamlining scale-up
Commercial Implications: Predicting market demand is never easy, particularly for emerging technologies that lack historic precedent. Manufacturing flexibility — both in scheduling and capacity for rapid production scale-up — is thus vital to the commercial success of the CTI.
Fortunately, because of the inherently disposable nature of cell therapy bioprocessing consumables, scale-up can be achieved while reducing capital expenditures and time burdens — e.g., compared with conventional monoclonal antibody (MAb) processes that often rely on inflexible, fixed apparatus. CTI manufacturing infrastructure capable of rapid and cost-effective response to changes in market demand could generate a greater annualized return on investment than conventional MAb facilities. For now, however, the MAb market is appreciably larger with 2010 US sales of $18.5 billion (14).
Unlike MAb manufacturers, whose significant infrastructural demands restrict flexibility in facility location, cell therapy manufacturers (beginning with single-use technologies) can build and operate facilities in areas with limited infrastructure, eventually including in less economically developed countries (LEDCs). In this regard, the CTI could act as a catalyst for positive social change. Process engineering has historically been a PhD-rich profession. But with the prospect of facility assembly in remote locations, it will be prudent to harness available labor by training resident workforces in standard processing techniques. That helps develop local skills and provide new jobs.
Cost of Goods
A vitally important aspect of all scaled-up therapeutics is to make them affordable to patients and healthcare providers. Future advances in bioprocessing technologies will undoubtedly provide step-change reductions in CoGs for cell therapies. But multiple opportunities already exist to minimize expenditures throughout clinical development, thus improving both profit margins and patient access. Manufacturing strategies that extract maximum value from existing expertise undoubtedly will position their parent companies favorably for commercial success.
One common change that both streamlines a process and helps to reduce CoGs is moving away from the traditional media-exchange protocols common in academic and research settings. Cell growth medium is typically changed every two or three days, an established process in laboratory cell culture that isn’t supported by data. By analyzing media consumption using common biochemical media analysis techniques, it is possible to establish consumption rates and thus develop a data-driven feed strategy for media replenishment based on empirical evidence.
During the first few days of culture, nutrient consumption is lower simply because fewer cells are present. As they approach confluence, their rate of media consumption rapidly increases. Often, by monitoring media consumption rates through multiple experiments, it is possible to devise a feeding p
rotocol that stipulates media replenishment less frequently during early culture stages and more toward the end of a process. This approach usually allows for complete removal of at least one feed to reduce the cost of an expensive raw material and labor associated with media changing (15).
Other approaches include making a richer basal media. Although this increases the cost per liter, the resulting improved cell growth kinetics reduce overall cost per cell upon harvest. It is also important to appreciate the impact of facility and labor on overall processing costs. Both are reduced considerably through media optimization.
Other major process changes relating to media composition include removal or reduction of undefined media additives and replacement or removal of xenosourced products, including fetal bovine serum (FBS). Such process changes reduce not only regulatory risk, but also supply chain issues associated with raw material sourcing (11). Xenosourced materials are associated with potential transmission of zoonoses. Equally important, few such materials come from scalable sources that can accommodate the amount of commercial supply associated with a blockbuster product (11).
Commercial Implications: As the scale of a cell therapy bioprocess increases, the relative costs of different aspects of that process — such as fixed overheads and consumables — will fluctuate. What began as a significant expenditure at research scale (e.g., facility costs for allogeneic cell therapies) may proportionately be very much reduced at industrial scale — and vice versa (e.g., QP inspection for autologous therapy). Media cost is invariably one part of the process that greatly increases in parallel with scale, making it difficult to realize significant economies of scale during early process planning. With a generic allogeneic mesenchymal stem cell product produced at 1,000 doses/year, for example, media accounts for 24% of total manufacturing cost, and facility cost is a much more significant 49%. However, for the same process scaled up to 100,000 doses/year, 58% of money spent on manufacturing is spent on media compared with only 15% on the facility (Table 2) (16).
Table 2: Changing proportions of cost of goods and scale (16)
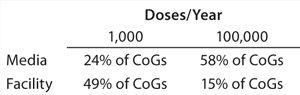
As that clearly demonstrates, although media optimization studies may seem expensive and unnecessary at smaller scales, they can lead to significant cost savings after commercialization. Furthermore, at larger scales, parameters used to measure cost-effectiveness are subject to change. Instead of cost per batch, for example, cost per million cells may become more relevant. With those vital thought processes incorporated early in development — as well as understanding process economies of scale — overall CoGs for an entire scaled production can be significantly reduced.
Commercial benefits of designing and executing a cost-effective cell therapy manufacturing strategy are arguably more complex than for conventional (bio)pharmaceuticals, principally due to the interdependency of product characteristics and biomanufacturing processes (8). Cell therapies shipped in a fresh (not cryopreserved) state particularly require efficient integration of manufacturing and distribution strategies to maximize potential market sizes and/or penetration — and thus the likelihood of commercial success.
The Way Forward
Cell therapy represents an extraordinary opportunity to provide life-changing treatments for millions of patients worldwide. The CTI bioprocessing community is charged with a bold and exciting task to deliver these novel and biologically complex therapeutics to patients safely, efficaciously, and affordably. Given the size of the potential market, the CTI’s opportunity is on a par with that of the more mature multinational pharmaceutical, biotechnology, and medical device industries.
For CTI developers to balance processing innovation with commercial realities, leading industry experts widely agree that the only route forward for cell therapy companies is minimizing capital commitment and staying virtual for as long as possible (17, 18). Such developers can maximize capital efficiency and the extent to which external expertise can be leveraged before locking down their final processing strategies. In addition to seeking the advice of experienced consultants, developers might want to engage with the growing network of cell therapy translation centers that have expertise in implementing clinical trials, including product manufacture.
Bioprocess technologies are constantly evolving, as is the commercial environment in which the CTI operates. Historically, bioprocessing has focused on optimization of existing technologies — in particular, refinement of existing platform processes (19). Such techniques are a pragmatic route for the first wave of cell therapy products — those currently in late-stage clinical trials — to get to market. Although changes discussed herein can significantly improve the maximum achievable manufacturing capacity of a process, however, ultimately such processes will be insufficient to meet future market demands. Incremental refinements in processing technologies thus will need to be succeeded by a series of ground-breaking techniques at every step in production and downstream processing to support long-term growth of the CTI (20, 21). For this industry to unlock its true potential, multiple integrated innovations for cell processing will need to be as radical as the series of “disruptive” technologies that facilitated the processing of electrons — from valve to transistor to silicon chip.
Author Details
Corresponding author Natasha L. Davie is a BRITS investigator for the Advanced Centre for Biochemical Engineering at University College London, and a researcher for The Harvard Stem Cell Institute and Harvard Medical School; Holyoke Center, Suite 727W, 1350 Massachusetts Avenue, Cambridge MA 02138; 1-504-4703, fax 1-617-525-4330; natasha_davie@harvard.edu. David A. Brindley is a BRITS investigator for the Advanced Centre for Biochemical Engineering at University College London and a researcher for The Harvard Stem Cell Institute and Harvard Business School. Emily Culme-Seymour is director of the London Regenerative Medicine Network, and Chris Mason is a professor of regenerative medicine bioprocessing at The Advanced Centre for Biochemical Engineering.Mason is principal investigator; and Davie, Brindley, and Culme-Seymour are investigators on the British Regen Industry Tool Set (BRITS) project funded by the Technology Strategy Board under their Regenerative Medicine Program: Value Systems and Business Modeling. The authors have no other relevant affiliations or financial involvement with any organization or entity that has a financial interest or conflict with the subject matter or materials discussed herein.
REFERENCES