Earlier this year, the FDA issued its long-awaited process validation guidance document, which had been several years in development. It is well written and effectively articulates what many progressive companies have been thinking and doing for years. But many people in the industry are asking questions.

Part 1 of this article described the history of process validation before the FDA’s quality by design (QbD) initiative and discussed QbD in general. It also described the new process validation paradigm according to the 2011 guidance in a QbD context. Part 2 concludes here with examples, a focus on postapproval activities, and general implications.
Two Examples
A Cell Culture Unit Operation: After expansion of frozen cells from vials in vessels of increasing size, you have sufficient biomass to express a protein in your production reactor (production phase). This step in the manufacturing process will express a batch of protein that will be subsequently recovered and purified to yield the BDS. It is important to clearly define the purpose of this step and the needs for each element of the process, which come directly from development work. For this discrete step, you must evaluate several elements.
First, identify the precise steps in this unit operation, clearly defining what is performed and why. Deciding to perform an activity at a discrete point in a process must be described with inclusion of its rationale and constraints within each action. In this particular phase, actively growing cells must be transferred and diluted into fresh medium while preventing both physical and chemical shock. The diluted cells grow for a period (with appropriate additions) to yield an active biomass that can be converted to production mode so that its energy can translate from biomass production to product expression. The culture state is monitored, and it is controlled through nutrient addition to a predefined titer before harvest.
Each of those actions is first carefully defined and described. Then you can move to the next component of your PV process.
There are two types of raw materials are used in production. Classical raw materials include components of cell culture or fermentation broth (a mixture of inorganic and organic materials that form the basal medium), and additional nutrients (e.g., a carbon source, acid-buffering material to maintain pH, and inducers). The quality of such materials is defined in official specifications used to purchase them, which should be derived from carefully designed experimentation. The second type of production raw material is the end product of a previous bioreactor (n – 1). That typically comprises host cells along with an appropriate genetic construct, grown up to sufficient biomass with appropriate physiological activity to yield the manufacturing requirement.
Table 1:
Figure 4 details of for the two examples described herein
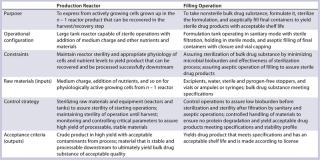
Through appropriate experimentation, the design space should have been defined already as part of your QbD program. To assure that this production process can be controlled within that space, development work has defined which parameters must be controlled within which ranges by defined control strategies. To attain sufficient yield, for example, you will have defined the feeding strategy of key nutrients, specifying precise amounts of nutrient to be added during each phase of production. Your definition might cover the carbon-source addition rate by feeding and dissolved oxygen (DO) levels that must be controlled through sparging rate or gas composition.
Harvest control strategies are different for products that are excreted into the cellular medium and those retained by cells. Intracellular products typically require harvest after lysis of cells. But to make subsequent steps easier and more reliable to perform with excreted products, you might define that you want no cell lysis. So the strategy would be to increase productivity to the point that cells begin to die but not lyse. That would prevent release of host-cell proteins that can be difficult to remove, including intracellular proteases that can degrade your product during recovery and purification.
Yield is not the only required output at this stage. Remember that the maximized output of this step is a raw material for the next steps downstream, which include harvest or recovery followed by purification. So although you want an economically high yield from your bioreactor, you also want material that can be processed through the next step without physical problems (such as clogging filters). And you want it to be chemically stable, with decreased tendency to degrade over the time needed for purification. So upstream output is defined by downstream requirements.
Critical control points are documented with acceptable ranges for controlling and monitoring them to ensure that products meet their CQAs. In addition, you should describe the less-critical process parameters to be monitored but not controlled to such a high level of precision.
Your PV protocol will define all those key elements of control and ranges as well as outputs that must be attained for downstream success. Some outputs are defined chemically, physically, biologically, and microbiologically. And in some cases, straightforward tests to measure success may be unavailable. You might thus need to have a small-scale version of the next step (or two) to measure the appropriateness of this unit operation’s output. The results become the measures of success of the PV study.
A Final Fill Operation: Because biologics cannot be terminally sterilized, they must be sterile filteredand aseptically filled into vials, ampules, or syringes. It is important to de
fine the purpose of this unit operation together with the rationale for all steps in a process.
Follow the same strategy here as defined for bioreactor production above. Each step is defined in a unit operation along with its purpose, constraints, and strategies. In this case, concentrated BDS must be diluted and the concentrations of active and excipient ingredients adjusted to the final DP formulation. Nonsterile material must be filtered, filled aseptically into final containers, then processed (lyophilized and stoppered or simply stoppered) before capping and packaging.
Raw materials at this stage comprise water and excipients, the container and closure, and the BDS (which is the output of the previous step). Purchased RMs or components are defined by specifications in your vendor agreement — or in the case of the water, by the appropriate pharmacopeial standards. The BDS is defined by specifications you have defined in your design-space development work with attention to CQAs.
Your filling-operation control strategy would have been developed through development work to address how your BDS is sterilized by filtration and to define the microbial limits that filtration process can handle. The strategy should also define exactly how to mix the product to homogeneity without causing shear stress on a product. And it should address stability of a product held until filled in a worst-case operating time frame. These are just examples of the many elements to define in this case.
Filling outputs are defined by product specifications as well as product stability under defined storage conditions. Other attributes may also be included. This is the drug product, itself — which is directly used by patients — so product CQAs are appropriate here.
After all unit-operation validations are complete, it is customary to link them together (at least from cell bank to BDS) and thus create one final demonstration of your whole process. The filling step often represents the final step as a stand-alone validation.
But that is not all there is to process validation. A battery of other studies must be performed, including those addressing media and buffer preparation (involving mixing studies and use tests). In addition, studies should account for cleaning, sanitization, and hold studies for media, buffers, and product and process tanks at intermediate manufacturing stages. Biologic–specific studies include viral removal and cell-line stability studies as well as column and filter multiuse studies, often performed at small scale in scaled-down operations. Under the new paradigm, I believe those will be required and performed similarly to how companies are used to approaching them, with large-scale confirmation of their conclusions.
The Postapproval Phase
In licensure with submission of a QbD package and just a few demonstration runs — or more likely a commitment to demonstrate the process for a defined number of runs (20–30, for example) after approval — the third phase of PV formally begins with that first defined number of batches. On completion of those batches, resulting data would be analyzed and submitted to the FDA along with confirmation that the process has worked as predicted within its design space. While the agency processes those data, you would be free to sell your product.
As a process is operated in commercial mode, data are continuously gathered and subjected to analysis with appropriate trend charting. At some point, a periodic review would be made that might coincide with the annual review. It’s more likely that changes would be made as trends are noted, issues arise, or new technology is made available. At that point, you would invoke your robust change-control process. Very similar to change processes that have been used so far, it would feed directly into the product history file that lays down the chronology of process and product changes by addressing each change with the rationale for it and the strategy for moving from one validated state to the next. That would entail a risk-based analysis of each proposed change against a backdrop of predefined design space, with an increase of knowledge gained through the operational phases after commercialization.
Making It Better
The three-phase activity (robust development, traditional PV, and monitoring) leading through the life of a product could bring us to a new paradigm in which process and product knowledge yield products that are developed under more discriminating conditions, with quality built into both them and processes that make them. Manufacturing processes will be better understood in the future so they can be accepted by the agency with higher confidence — and thus approved quicker with less concern. Improved understanding should lead to more ruggedly controlled processes with improved reliability and higher success rates in operation than ever before. The industry can earn the ability to manipulate process parameters with higher confidence of their outcomes and less oversight by regulators. By continuously monitoring these processes, companies will be able to predict drift and make adjustments that keep their processes (and outcomes) under even better control before they have an opportunity to fail. So the concept of continuous improvement can be incorporated into daily operations. Getting to that state requires up-front investment of resources, but the payback should be very good over the long term. PV will become a truly value-added activity.
How might all this affect timelines, up-front costs, and pathway for biopharmaceutical product and process development? Under this new paradigm, more process knowledge will be needed at the time of submission than most companies currently possess. Some might choose to begin process development for their commercial processes as before (during phase 2 clinical trials, when positive results suggest success) and simply extend their timelines so that submission occurs later. But I have never seen senior management accept such an option before.
That leaves a company with two options. It can begin this work earlier (at the riskier stage of phase 1), starting PV without clear indication of safety or efficacy. Most companies probably will continue to begin PV studies at phase 2 and maintain their submission dates, but simply invest in more resources to complete the studies on time. Doing so will require strategies and risk analyses to dovetail all interrelated studies — including upstream, downstream, and fill–finish work and stability studies — into the short time frame.
If the industry embraces this QbD–PV concept, what happens to the old requirement of “three PV runs and done?” Clearly that does not provide a large degree of assurance that a process is good or has been successfully demonstrated. So will the agency start requesting 20–30 runs before product approval? I think not — and hope not! With better understanding of a process and its controls in the development phase based on QbD and PV, companies should be better able to demonstrate linkages of inputs and process parameters to outcomes. So the number of runs needed might actually decrease to zero for licensure. Instead, companies might end up with a commitment to monitor their processes intensively from initiation through perhaps the first 20–30 runs after product approval. They would also commit to monitoring these processes throughout the product life-cycle and make adjustments accordingly. Results of such monitoring would become another set of parameters to examine during each annual product review.
Author Details
Peter H. Calcott, PhD, is president of Calcott Consulting, 931 Mendocino Avenue, Berkeley, CA 94707; 1-510-585-8256; peterc@calcott-consulting.com; www.calcott-consulting.com.