Single-use technology is being examined for implementation in an increasing number of steps in the biopharmaceutical manufacturing process. Some examples of currently available disposable components include filter capsules, tubing, connectors, and biocontainers (for storage, mixing, and bioreactors), as well as devices for chromatography and multipass tangential-flow filtration (1,2). This technology was first implemented in upstream and API downstream processes such as media and buffer preparation, followed by upstream bioreactors and mixers (3). The single-use trend has most recently shifted further downstream toward sterile formulation and filling operations (3). Vaccine manufacturers in particular are using disposable systems to streamline their final filling processes.
Widely recognized benefits of single-use systems in biopharmaceutical manufacturing processes include elimination of cleaning/sterilization and associated validation; decreased operator exposure to product streams; and reduced installation, assembly, and maintenance costs (4). Improved productivity and reduced environmental footprints are additional benefits of single-use technology over traditional stainless steel systems for bioprocessing (1).
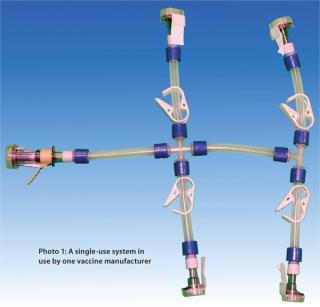
A unique characteristic of the final filling step is that it has no additional filtration step downstream. Consequently, this operation has generated some concern regarding potential contamination of a final product by particulate matter and endotoxins. Here we discuss an approach for monitoring particulate and endotoxin content in single-use systems for a final filling application.
Regulatory Landscape
The current regulatory landscape is such that no US FDA, EU EMA, Japan MHLW, or WHO regulations or guidelines are specific to particles in process equipment such as single-systems, final filters, filling equipment, final bulk drug containers, or even drug-product dosage containers before filling. Existing regulations must be examined in more detail as they apply to drug manufacturing to determine how they relate to regulatory requirements for particles in finished drug products.
Process equipment itself does not fall within the scope of drug regulation. All existing drug manufacturing regulations and guidelines for particulate quality apply to external cleanroom environments or finished drug products after filling in their final dosage containers. In addition to inspection of final filled dosage units for visible particles (according to USP chapter ), finished injectable drug products are also subject to lot sample testing for microscopic particulates (e.g., according to USP chapter ) (5,6). For global harmonization, such tests were incorporated into ICH Q4B (7).
The potential impact on the quality of final drug product from process equipment when it comes into contact with final bulk drug formulation is recognized by regulatory authorities. But the only specified requirement for process equipment (e.g., filters, connectors, filling lines, and bulk drug containers) is described in US 21 CFR Part 211.65(1), which states, “Equipment shall be constructed so that surfaces that contact components, in-process materials, or drug products shall not be reactive, additive, or absorptive so as to alter the safety, identity, strength, quality, or purity of the drug product beyond the official or other established requirements” (8). A similar statement can be found in EU GMP Annex 1: “The parts of the production equipment that come into contact with the product must not be reactive, additive or absorptive to such an extent that it will affect the quality of the product and thus present any hazard” (9).
Those GMP regulations and guidances do not specify particulate quality of process equipment, but rather they only recommend that drug manufacturers ensure that their process equipment does not adversely impact the quality of their final drug products. So users must establish reasonable controls to ensure that their process equipment does not cause drug products to fail its particulate quality specifications.
Injectable drug products must be “essentially free of visible particles.” USP chapter states, “Each final container of all parenteral preparations shall be inspected to the extent possible for the presence of observable foreign and particulate matter (otherwise termed ‘visible particulates’) in its contents. The inspection process shall be designed and qualified to ensure that every lot of all parenteral preparations is essentially free from visible particulates” (5). Similarly, EP 2.9.20 states, “Injectable solutions, including solutions constituted from sterile solids intended for parenteral use, should be essentially free from particles that can be observed on visual inspection” (10).
Observation of a “visible particle” during inspection depends on drug and container clarity, lighting, particle size, optical density, refractive index, color and contrast, and operator or automated inspection sensitivity and reliability. “Essentially free” is not defined. Only those individual drug-product containers observed to contain visible particles are subject to rejection, so limits can vary for the number of unacceptable drug product containers with one or more visible particles per batch that can be rejected to meet an interpretation of “essentially free of visible particles” for released dosage units. The regulatory expectation is that “essentially free of visible particles” refers only to released final dosage units.
Single-use technology users have expressed concern regarding the endotoxin content of disposable systems. As is the case for particles in process equipment, no regulations or guidelines are in place for endotoxins in such equipment. The recognized assay for bacterial endotoxin — the Limulus amebocyte lysate (LAL) test — is used to detect or quantify bacterial endotoxins that may be present in or on an article sample by testing an appropriate liquid rinse volume. Details of that test are found in USP (11) and EP 2.6.14 (12).
Single-Use System Examples
Manufacturing processes that produce single-use systems are typically manual operations. It is meaningful to consider both the nature of the components in such systems as well as the quantity of connection points involved. Potential sources of particulate contamination include the nature of each component as well as the cleanliness of hoppers or bins, the air quality of the systems’ manufacturing environment, and the manufacturing process itself. A key potential source of particulate contamination during single-use system manufacturing may come from technicians cutting tubing and affixing tubing lengths onto the barbs of tubing connectors. Although the assembly of disposa
bles is typically performed in cleanrooms with strict gowning procedures, skin contact of operators with these systems can be a major source of bioburden and endotoxins (4).
The greater is the quantity of connection points on a system, the greater will be the amount of manipulation that system receives from an operator during manufacturing — and perhaps the greater will be the likelihood of particle and endotoxin contamination introduced by operators. So Pall chose tubing and connector suppliers to provide components for Allegro single-use formulation and filling manifold systems on the basis of
- a robust ISO 9001 quality system that includes full traceability records
- an ability to supply components in USP and qualified pharmaceutical-grade resins
- appropriate clean manufacturing facilities
- supplier agreements that ensure quality, consistency of supply, and change control.
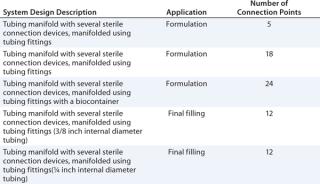
Table 1 lists examples of single-use systems currently produced by Pall for vaccine formulation and final filling applications.
Table 1: Single-use system designs for use in biopharmaceutical or vaccine formulation and final filling processes
Testing Protocols
We evaluated two unique Pall Allegro single-use system designs for filling applications. One system from every lot manufactured with each of the two single-use systems was evaluated over a period of 22 months, for a total of 32 samples. The method is detailed below. In summary, we dispensed LAL-negative purified water into each system. Then we agitated each system for a period of time before draining the water and evaluating for particulate matter and endotoxin content. Our assays were consistent with USP chapters and , respectively.
We considered the particulate matter of the water rinse sample from a single-use system to represent the total particles present that would come out in initial fluid-contact void volume without preflushing. We considered the endotoxin content of the water rinse sample from each single-use system to represent the concentration of endotoxin present in the first void volume without preflushing. Our objective was to generate endotoxin and particulate data for single-use systems and demonstrate that they can be evaluated in a quality control (QC) manner to determine suitability for use — particularly in a filling application.
We compared the performance of each system in each test with the following reference standards:
- Particulate matter: The limit is defined as ≤3 particles/mL of ≥25 µm from one system rinse volume when tested according to the microscopic particle count method and limit stated in USP chapter for small-volume parenterals.
- Endotoxin content: USP chapter based on the water-for-injection (WFI) standard described in the monograph.
Protocol Detail — Generation of Single-Use System Water Samples: We used LAL reagent water from Charles River Laboratories in Charleston, SC (hereafter referred to simply as “water”) to rinse the systems. It is known to be both LAL-negative and low in particulate matter. Water was prefiltered and dispensed into each system such that it could freely move throughout the system. After agitating a system for one hour, we drained the water and divided it into two volumes: a 5-mL sample taken for endotoxin testing and the remainder designated for microscopic/visible particulate matter testing.
Protocol Detail — Endotoxin Determination: We evaluated water samples from the single-use systems for endotoxin content using a method consistent with the photometric techniques section of USP chapter . The endotoxin test protocol uses a standard LAL kinetic chromogenic method that relies on the ability of lysate from amebocytes that come from horseshoe crab blood to clot in the presence of endotoxin. A set of known endotoxin concentrations (“standards”), the test samples, and positive product control (PPC) samples (samples with added known concentrations of endotoxin) are mixed with LAL reagent to induce clotting. We measured the time required for each sample to clot using a spectrophotometer and plotted a curve of the standards. Once the endotoxin concentration of the PPC samples confirmed the absence of inhibitory or enhancing effects of the sample diluent, we interpolated the endotoxin content of a test sample from that standard curve and reported the values in endotoxin units per milliliter (EU/mL).
Protocol Detail — Particulate Matter Determination: We evaluated water samples from each single-use system for particulate matter using a method consistent with the appropriate sections of the microscopic particle count test procedure of USP chapter . The particulate test protocol uses a microscopic optical particle-counting method. Liquid samples are gravimetrically filtered through a black gridded recovery membrane, which is counted for particles in ranges specified in USP chapter . Values are reported in particulate matter counts per mL (particle size ≥25 µm). We also observed those recovery membranes for presence of visible particles.
Results and Discussion
For all 32 samples we tested, the particulate matter and endotoxin content were within acceptable limits set for each test. Controls for each test behaved as expected: The LAL reagent water neither enhanced nor inhibited endotoxin in the samples, and the test environment we used for measurement of particulate levels was acceptable. Table 2 summarizes our results.
Table 2: Results of particulate and endotoxin content testing of single-use systems used in a final filling application (evaluated over a 22-month period)

Our study results demonstrate that Allegro single-use tubing manifolds are manufactured in a manner that is consistent with regard to particulate and endotoxin levels in rinse volumes, as indicated by the consistency in results of the assays presented here. The assembly process does not contribute endotoxin, and the systems were shown to contribute particulate matter ≥25 µm into void
volume rinses within acceptable limits for injectables detailed in USP chapter . The absence of visible particles in these systems, along with the low levels of detected subvisible particles, suggests a lower risk to patients. It is worthwhile to note that although no visible particles were detected in these systems (as per USP chapter ), parenteral preparations should be inspected and found to be “essentially free from visible particulates.”
The results from this study, together with data generated for single-use systems in formulation applications, led us to consider evaluating a larger, more complex system that could be considered a “worst-case” assembly of components. This would serve as a “master product” representing most tubing manifold design variants for simplified QC purposes. Particulate matter and endotoxins in the fluid paths of single-use final formulation and filling tubing manifold systems can be monitored using compendial methods. Limits can be applied to a QC test rinse solution, with sample volumes based on the rinse volume of a simulated product (single-use system).
DESCRIPTION OF THE PALL MASTER SIMULATED PRODUCT
Single-use manifold system model containing several loops of tubing lengths, an Allegro biocontainer, Kleenpak sterile connectors, multiple types of tubing fittings, and ~13 ft of tubing (total): 62 connection points
Although no specific standard is available for assessing particles from process equipment, a standard for sterilization of healthcare products (AAMI/ANSI/ISO 11137) describes assessment of products for bioburden, which effectively equates to viable “particles.” According to AAMI/ANSI/ISO 11137-2, a master “simulated product” can be considered representative of multiple products (e.g., single-use tubing manifold systems). It must
- contain identical or equivalent components and materials to those used to build customer designs, though varying in size and quantity
- be constructed of all main component types and materials to represent the common single-use subcomponents
- be manufactured using the same procedures in the same facility as customer orders
- be packaged in the same manner and with the same materials as are used for customer orders.
In addition, to be applicable to formulation and filling manifolds, a system should be designed to incorporate multiple connection points and simulate a complex manifold like those requiring the most handling during manufacturing and assembly. These characteristics of the simulated product allow it to be considered an overestimate (“worst-case”) for the particulate matter and endotoxin content of products Pall manufactured for any custom manifold order. The data periodically generated for simulated product systems demonstrate that all related single-use manifold systems continue to meet these particulate and endotoxin limits.
Master Simulated Product Definition, Test Design, and Results: The master simulated product contains multiple components and was designed to contain a very high number of connection points. This increases the number of tubing cuts and degree of required manipulation during system assembly and includes as many different components as possible to represent a wide range of components found among single-use manifold system customer orders.
Design of the master simulated product does not contain filter capsules or biocontainers because such components are controlled and assessed separately for particles and endotoxins. Pall sterilizing filters are preflushed in manufacturing and packaged in a class 10,000 cleanroom. Flush or soak effluents of lot samples are QC tested for particles and endotoxins, respectively. Allegro biocontainers are also manufactured in a class 10,000 cleanroom, with sample rinse volumes tested in a similar manner for acceptable quality levels. Particulate and endotoxin test results for Pall filter capsules and Allegro biocontainers are provided in their respective validation guides, and lot sample test results are certified to meet compendial limits for particulates in injectables and for endotoxins in WFI. The “Description” box summarizes Pall’s master simulated product.
We prepared nine master simulated single-use system samples, three from each of three lots produced. Samples were evaluated using the rinse-and-recovery procedure with LAL reagent water described above, and water recovered was evaluated for endotoxins and particulates as mentioned above. We compared results with the same limits for both. Results were acceptable to the above-mentioned limits. Additional samples of the master simulated product will be built and evaluated as well. Table 3 details the results.
Table 3: Quantification of endotoxin and particulate matter determination in the “simulated product” single-use system
When single-use systems are used in the final filling stage of biopharmaceutical product manufacturing, concerns regarding potential contamination of product fluid streams by particulate matter and/or endotoxins are reasonable. This requires careful study. A multipronged approach includes proper vetting of subcomponent suppliers, in-house inspection procedures, with a QC approach to finished products as discussed here. Together, those can help a company create a knowledge base of information to develop a robust monitoring program for single-use systems.
Author Details
Donna Riedman is a senior technical specialist in the scientific laboratory services department, and Jerold Martin is senior vice president of scientific affairs at Pall Life Sciences, 2200 Northern Boulevard, East Hills, NY 11548; 1-516-801-9546; donna_riedman@pall.com
REFERENCES