Where were you in 1987, and what were you doing? I’m not too embarrassed to say that I was beginning my last year of high school and paying far more attention to guitar lessons and writing my first novel than what I might eventually do for a career. Meanwhile, the US FDA was publishing a guidance document on process validation that the biopharmaceutical industry has relied on ever since. I’m willing to bet that quite a few readers of this issue weren’t yet working in the industry at that time either — and one or two could even have been in the New England crowd with me at a Def Leppard concert that year.
A lot has happened since then in the United States: four different presidential administrations dealing with wars and major economic boom-and-bust cycles, a huge federal budget deficit turned into a surplus and then into an even larger deficit, the growth of the Internet from a niche military/academic application to a ubiquitous and vital personal and business tool, and the dawn of a new millennium. Biopharmaceutical manufacturers saw the end of the establishment license (ELA) and product license application (PLA) processes, the rise of the biologics license application (BLA), the birth of the well-characterized biologic, dozens of product approvals (1), and the coming of biosimilars. All the while, your main source for answers about the FDA’s expectations regarding process validation has stayed the same. It was all about testing, instrument qualification, and process robustness and repeatability. Now finally, as of January 2011, the agency has finalized an updated guidance (2) to bring process validation into the modern era of risk management and quality by design (QbD).

It is only fitting, then, that the primary focus of IBC’s 15th annual “Process and Product Validation” conference (part of “Biopharmaceutical Development and Production Week”) in Bellevue, WA, 14–15 March 2011 was the similarities and differences between this new guidance document and the one it replaces, with which everyone should by now be very familiar.
FDA Session
During the Monday morning plenary session on 14 March (shared with IBC’s sixth annual “Technology Transfer for Biopharmaceuticals” and seventh annual “Outsourcing Manufacturing of Biopharmaceuticals” meetings), three FDA representatives literally phoned in their teleconference presentations on the Office of Biological Products (OBP) pilot program for QbD (Patrick Swann, OBP’s deputy director), the concept of continuous verification (Grace McNally, consumer safety officer at the Center for Drug Evaluation and Research’s division of manufacturing and product quality), and risk-based approaches to process understanding and control (Mansoor Khan, director of CDER’s division of product quality research). Anyone who attends a west-coast conference has come to expect less FDA participation than can be had on the east coast, so their long-distance session was a welcome addition to the program.
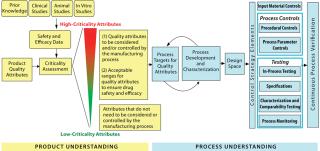
Pilot Program: The OBP launched its QbD pilot program for biotech products in July 2008 with two streams, one for new molecular entities at the investigational new drug (IND) or BLA stage and the other for postapproval-stage molecules. The pilot program has provided an opportunity for the biopharmaceutical industry and the FDA to evaluate and identify best practices for key QbD elements of target product profiles, critical quality attributes (CQA), risk assessment, process characterization for design-space definition, CQA-focused control strategies, and expanded change protocols. This builds on the concept of well-characterized biologicals (a.k.a. specified biologics), which came about in the late 1990s when it was recognized that analytical methods had improved to the point at which biologics could be analyzed and described well enough to separate their identities from their processes, at least somewhat (3,4,5). A few years later, CDER took over responsibility for those well-characterizable products from the Center for Biologics Evaluation and Research (CBER).
OTHER GUIDANCE DOCUMENTS
The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) brings together regulatory authorities and pharmaceutical companies from Europe, Japan, and the United States to discuss scientific and technical aspects of drug registration. Since its inception in 1990, ICH has worked through its global cooperation group to harmonize the increasingly global face of drug development. Its mission is to help ensure that safe, effective, and high-quality medicines are developed and registered with the most efficient use of resources. The organization focuses on four main subjects: quality, safety, efficacy, and multidisciplinary guidelines, which include the MedDRA standardized medical terminology guide and the common technical document (CTD), a format for submitting information for regulatory review in all participating countries. ICH celebrated its 20th anniversary in 2010 (www.ich.org/fileadmin/Public_Web_Site/News_room/C_Publications/ICH_20_anniversary_Value_Benefits_of_ICH_for_Regulators.pdf). In relation to process validation, four ICH subtopics are most relevant: Q8, Q9, Q10, and Q11.
ICH Q8(R2): Pharmaceutical Development was adopted by the European Union in June 2009, by the United States in November 2009, and by Japan in June 2010, after having been finalized in November 2005. This document is intended to provide guidelines for drug products as defined in the scope of Module 3 of the common technical document (CTD), which is ICH topic M4. The guideline does not apply to contents of submissions for drug products in clinical research, but its principles are important to consider during that stage. An annex to the tripartite harmonized ICH text was finalized in November 2008 and incorporated into the core document, which was then renamed Q8(R1). That annex provided further clarification of key concepts and described the principles of QbD. It showed how concepts and tools (e.g., design space) outlined in the parent document could be put into practice. When a company applies QbD and quality risk management (ICH Q9, see below) as part of a pharmaceutical quality system, opportunities arise to enhance scienceand risk-based regulatory approaches (as described in ICH Q10, see below). The Q8 guideline was revised to (R2) in the summer of 2009 to reflect minor corrections, and it can be downloaded online at www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q8_R1/Step4/Q8_R2_Guideline.pdf.
ICH Q9: Quality Risk Management was adopted by the European Union in January 2006, by the United States in June 2006, and by Japan in September 2006 af
ter the tripartite harmonized ICH guideline was finalized in November 2005. This guideline provides principles and examples of tools for quality risk management that can be applied to all aspects of pharmaceutical quality including development, manufacturing, distribution, and inspection and submission/review. It can be applied throughout the lifecycle of drug substances and medicinal products, biologicals, and biotechnological products and covers the use of raw materials, solvents, excipients, packaging, and labeling materials. The document can be downloaded at www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q9/Step4/Q9_Guideline.pdf
ICH Q10: Pharmaceutical Quality System was adopted by the European Union in July 2008, by the United States in April 2009, and by Japan in February 2010 after the tripartite harmonized ICH guideline had been finalized in June 2008. This document applies to pharmaceutical drug substances and drug products (including biotechnology and biological products) throughout their lifecycles. Companies should apply its elements appropriately and proportionately to each stage. The guideline can be downloaded online at www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q10/Step4/Q10_Guideline.pdf.
Other Documents: Real-world experiences of companies and regulators with the Q8, Q9, and Q10 guidelines made ICH aware of a need for some clarification of key issues. The latest version of its resulting questions-and-answers document was finalized in November 2010 and can be downloaded online at www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q8_9_10_QAs/Q-IWG_QAs_Step4/Q8_Q9_Q10_Question_and_Answer_R4_step_4_November_2010.pdf.
ISPE has published a guidance document that’s free for its members and available to nonmembers at a nominal cost: ISPE Product Quality Lifecycle Implementation Guide: Overview of Product Design, Development and Realization — A Science-and Risk-Based Approach to Implementation. International Society for Pharmaceutical Engineering: Tampa, FL, October 2010; www.ispe.org/ispepqliguides/overviewofproductdesign. According to ISPE:, the guide is “the first in a series of product quality lifecycle implementation good practice guides that will describe enhanced, QbD approaches to product realization and is an introduction to and an overview of the guides series.” To address product and process development, transfer to and establishment of commercial manufacture using science-and risk-based approaches, the series will cover critical quality attributes (CQAs) and critical process parameters (CPPs), design space, and control strategy.
In addition, ASTM International has published a standard that supports ICH Q8 and Q9 titled ASTM E2537-08: Standard Guide for Application of Continuous Quality Verification to Pharmaceutical and Biopharmaceutical Manufacturing. According to ASTM, “The accumulated product and process understanding used to identify critical quality attributes (CQAs), together with the knowledge that the risk-based monitoring and control strategy will enable their control, should provide confidence to show validation of each batch manufactured — as opposed to a conventional discrete process validation effort.”
ICH Q11: Development and Manufacture of Drug Substances was endorsed as a topic by the ICH Steering Committee in April 2008. This new guidance is proposed for active pharmaceutical ingredients (APIs) harmonizing the scientific and technical principles relating to the description and justification of the development and manufacturing process (common technical document sections CTD S2.2–S 2.6) of drug substances including both chemical and biotechnological/biological entities. The document is only at stage 1 of the ICH process currently, and a concept paper can be found at www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q11/Concep_Paper/Q11_Concept_Paper.pdf.
The OBP pilot program offers individualized examination of QbD initiatives submitted in market applications for biotech therapies for a number of original and supplemental biologic license and new drug applications. Manufacturers — such as Genentech, an early program participant — voluntarily provide chemistry, manufacturing, and controls (CMC) information in an expanded change protocol describing their implementation of QbD and risk management. Their candidate products are monitored by trhe OBP throughout product development and testing after early discussions with FDA reviewers about R&D issues.
For the pilot, the Office of Compliance (OC) will be part of review communication, so there is a need to transfer information among OBP, OC, and the field. Ideally, product reviewers should be present at initial QbD-type inspections. The Office of Compliance will play a key role in understanding the role of quality systems in QbD filings and their control strategies. (6)
Continuous Verification: According to Swann, the new guidance describes three stages of process validation during the lifecycle of a drug product, which falls into line with ICH Q8 (see the “Other Guidance” box). During early product and process development, process design builds criteria for testing, qualification, and setting specifications later on. “The commercial manufacturing process is defined during this stage based on knowledge gained through development and scale-up activities” (2). Process qualification encompasses many validation concepts familiar to those who have been working with the previous guidance document all along: Manufacturing equipment, tooling, and instrumentation, and utilities must be qualified using standard validation protocols along with an associated validation master plan, risk assessment, and requirements specifications. “During this stage, the process design is evaluated to determine if the process is capable of reproducible commercial manufacturing.” (2).
Continued process verification, the final stage, was the focus of Grace McNally’s presentation. “Ongoing assurance is gained during routine production that the process remains in a state of control” (2). McNally emphasized the “life-cycle approach” that makes process validation an ongoing activity — not something a company can do and then move on. “Criticality” (as of quality attributes) is a continuum, she pointed out, not an either–or question. McNally also pointed to several other sections of the Code of Federal Regulations that can help those working to implement these concepts into their manufacturing process development: 21 CFR Part 100a, 110a, 110b, 160b3, 165a, 165d, 180e, 211.42, 211.63, and 211.68.
Panel Discussion
After the FDA presentations, we followed session chair Victor Vinci (director of purification development and viral safety in bioproduct R&D, LRL, at Eli Lilly & Co.) to a smaller ballroom for process validation strategies and case studies from Acceleron Pharma, Amgen, Genentech, Hospira, MSD Biologics, and Pfizer. The next day — along with sessions covering analytical methods validation
and transfer and drug product validation from the American Society for Quality, Amgen, Centocor (Johnson & Johnson), Genentech, Hyde Engineering, Novartis, Pfizer, and ProBioGen — we were treated to a fascinating panel discussion with Vinci, Robert Repetto (Pfizer’s director of external affairs), and consultants Peter Watler (Hyde Engineering and Consulting) and Peter Calcott (Calcott Consulting).
Calcott said that process validation is transitioning “from a check-box activity to value-added.” Everyone emphasized the interplay between raw materials, process controls, and final outcomes (drug products). The new guidance documents what thought-leaders have talked about for a few years now: the team-oriented, lifecycle approach linking QbD and product/process development with risk assessment. Calcott described a “four Ds” approach: design (for standard requirements), demonstrate (by experimenting), document (using good science and good manufacturing practice, GMP), and determine (with ongoing monitoring). The old DQ/IQ/OQ/PQ approach doesn’t demonstrate process; equipment function is barely half the story. There are raw materials and inputs, process controls, and product attributes associated with every unit operation in a manufacturing process.
Repetto told us that mature companies have in recent years done “a pretty thorough job” of integrating risk management and QbD into their process validation activities. But not every company can employ dozens of statisticians and risk experts so may be less experienced with the new science and risk based approach. The onetime rule of thumb for running three verification lots is no longer so straightforward; it depends on the level of product and process knowledge the company has acquired from development studies.
“Is this a new paradigm?” Watler asked. “I hope it is; we really do need one.” He and others emphasized that three lots run early in the lifetime of a product cannot be expected to represent its process a decade later — or beyond. The new guidance document is not prescriptive; it is a framework for conversations between companies and the FDA, as well as among departments and colleagues within those organizations. Caldwell warned that such conversations, however, can end up pushing product licensing later as the agency asks for more information — unless a company begins with the necessary data and understanding to keep that from happening. “The proof of the pudding is not in the first three bites,” Calcott emphasized. “It’s eating the whole thing.”
A company must be able to adequately explain and defend the choices that it makes. “It’s the data,” said Repetto, “not the number of lots.” For some, three lots may be enough; for others, thee may not be anywhere near enough; and for companies with a great deal of process knowledge and understanding about its platform process three runs may be more than is necessary. “I can foresee a day when routine periodic oversight demonstrating ongoing process control, could become a more valuable tool to the agencies than coming in to inspect everything at one time.
An audience member commented that such an approach seems to lend itself well to products that aren’t scaled very large (e.g., personalized medicine and higher-titer production, high-concentration formulations), for example with the commercial scale similar to that of clinical trial material production. “For this approach to work you need development data that is representative of your full scale process. If that can’t be done for a unique production system you will still need to demonstrate process control, so your process validation plan would reflect the lower level of process knowledge. ” said Repetto. As companies move forward, they’ll have to be looking at the overall process picture through trending and base their decisions on data. The A-Mab case study published by CASSS was identified as a good example of modeling for predictability (7).
“I think the agency can get past having things set in stone,” said Calcott, and let companies use data and science to explain why they shouldn’t be right away. Experience at large scale shows that things may need adjusting as time goes on. Those working with platform technologies — e.g., monoclonal antibodies (MAbs) — get a head start because they have lots of data to begin with.
What about continuous verification? That’s a major focus of the new guidance. If a process changes over time, will its critical quality attributes (CQAs) do so as well? For now, there’s no easy answer. “How we choose CQAs will be in flux as we figure out how to interpret these guidances,” said Watler. (ICH Q11, “Development and Manufacture of Drug Substances” is only at the first stage of international work, so it still has a ways to go.) If an opportunity to change a process arises (e.g., biosimilar competition) a company will certainly want to revisit everything. “All the knowledge you gather with QbD helps you make those decisions,” Watler added.
But Calcott emphasized, “Don’t gather data for data’s sake.” There should be a purpose behind annual product reviews, trend charting, and other scheduled reviews; companies shouldn’t continually “tweak” their processes based on every little issue. Only real problems should be addressed with process changes: Remember, you’ll have to justify these things to regulators with good science and common sense.
“As we learn more about these products and molecules,” Repetto said, “we’ll know better how to proceed.” And Calcott pointed out that the very nature of a CQA as critical makes it important to investigate every associated failure and issue related to it. Over time, however, “clinical experience may lessen the importance or criticality of some CQAs.” In the early days of process validation, Repetto reminded us, a company might never know or care whether something shifted within specifications. With continued process monitoring, “it may be something you want to look at.” By studying trends over time, companies may be able to discover real problems earlier than they might have otherwise — early enough to prevent process failures, product losses, and expensive mistakes.
The Importance of Analytical Methods
After the “Process and Product Validation” final sessions focused on analytical methods validation and transfer, IBC’s inaugural “Analytical Technologies for Biopharmaceutical Development” meeting packed a small hall on 16–18 March, often with some attendees standing in the back of the room. With the new process validation paradigm, it’s never been more apparent just how vital process analytical technologies (PATs), statistical design of experiments, bioassays, and data management are to the success of a biotherapeutic product in development. None of the defining, evaluating, trending, and monitoring described in the new guidance — and all the other documents relating to QbD — can happen without knowledgeable analytical personnel using dependable equipment to perform robust and reproducible la
boratory methods behind the manufacturing scenes.
A presentation from Keith A Davis, a busy senior scientist from Pfizer’s bioprocess characterization and analytical support laboratory in St. Louis described several technological advances that have improved productivity and the quality of data his laboratory can gather. Cross-trained groups of analysts such as Davis described are increasingly tasked with supporting development of robust manufacturing processes. Among the vendors and instruments he thanked for stepping up to help them succeed are
- the Octet platform from ForteBio “for accurate, rapid MAb titer determination,” which formerly involved protein A high-performance liquid chromatography (HPLC) or enzyme-linked immunosorbent assays (ELISAs)
- ProA PreDictor plates from GE Healthcare for MAb sample preparation using protein A purification
- the Processor Plus automated gel staining and blot processing system, also from GE Healthcare
- the iBlot dry blotting system from Invitrogen
- Nanodrop spectrophotometric products from Thermo Scientific
- Kingfisher-96 systems (also from Thermo Scientific) for automated DNA extraction and qPCR analysis
- HPLC systems from Agilent Technologies for amino-acid analysis
- The BioScale ViBE bioanalyzer for host-cell protein and protein A quantitation
- SpotFire data management software.
Critical quality attributes, as Paul Motchnik of Genentech described in his presentation the morning of Wednesday 16 March 2011, are identified through analytical testing with a solid basis in platform knowledge and scientific literature. Tools such as multidimensional chromatography (discussed by Methal Albarghouthi of MedImmune), hydrogen-deuterium exchange with mass spectrometry detection (described by Steven Berkowitz of Biogen Idec), isoelectric focusing (explained by Brian Hosken of Genentech), miniaturized immunoassays (presented by Mats Inganäs of Gyros AB in Sweden), microchip assays (addressed by Xiaoyu Chen of Novartis), laboratory automation (the focus of presentations by Martin Vanderlaan of Genentech and Susanne Demarco of Pfizer), and new-era mass spectrometry (reported on by several speakers to close out the Analytical Technologies meeting on Friday 18 March 2011) are helping companies answer the questions regulators will ask when it comes time for product review.
A New Way of Thinking: When I worked for BioPharm magazine in the late 1990s, I remember numerous discussions of the confusion over terminology: The word validation only applied to processes, whereas qualification applied to instrumentation, and neither would apply to products themselves. Design qualification (DQ), installation qualification (IQ), operational qualification (OQ), and performance qualification (PQ) — none of these appear in the new guidance. Repetition of the phrases process qualification and product validation illustrates the fact that in many ways, this is a whole new world.
Peter Watler told me after the meeting that he heard Grace McNally make a specific point that the IQ/OQ/PQ/DQ terms were industry terminology rather than the FDA’s. “I have certainly heard folks make an issue out of the terminology,” he said, “which is really quite silly. The focus should be on what a study says, not about what it is called. And the FDA is clear on this. This has caused some in industry to divert validation efforts away from a scientific understanding to strict adherence of terminology and protocol. Fortunately the new guidance does away with that nonsense so industry can put resources into science and understanding rather than protocol and definitions.”
As always, the first place you should go is to the guidance document itself. But we hope to offer up some expert advice in our pages over the months (and years) to come.
Author Details
Cheryl Scott is senior technical editor of BioProcess International.
REFERENCES