Life-science companies that adopt “quality by design” (QbD) into their overall operations are expected to achieve the “desired state” of manufacturing. So concludes the Q10 document from the EMEA, US FDA, and the International Conference on Harmonisation of Technical Requirements for the Registration of Pharmaceuticals for Human Use (1). The ability to achieve an appropriate quality outcome must be designed into each manufacturing process rather than companies relying on final product testing. An increased focus on QbD ultimately requires manufacturers to make larger investments earlier in the life cycle of their products — during process development — well in advance of approved commercial operations. The goal is to develop a sound scientific basis for a manufacturing “control space” that accommodates a range of defined variability in commercial process materials and operations while continuing to produce desired product quality outcomes for bottom-line business benefits.
Today’s new regulatory environment highlights innovative ideas regarding process development and manufacturing and forces us to think about the practicalities of implementing them. To achieve QbD, manufacturers need to carefully consider the role of five important areas: process understanding, design-space development, design for manufacturing, process improvements, and process upsets.
Process Understanding Requires CollaborationSuccessful approaches to real-time quality assurance (RTQA) require that critical process parameters (CPP) driving variability in the critical quality attributes (CQA) be identified and understood during process development. They can then be measured and controlled in real time while each batch is being manufactured. This is what the FDA means by “process understanding.” It requires
-
a culture of continuous improvement that produces sufficient process understanding so that regulatory approval is not needed for process changes
-
close collaboration between process development and manufacturing teams
-
deployment of appropriate enabling technologies.

WWW.PHOTOS.COM
Collaboration can drive QbD. Advantages include adoption of better practices and sustaining business benefits such as higher levels of process predictability and quality compliance across a global manufacturing network.
Figure 1 shows the basic elements of process development and manufacturing collaboration. As shown, the level of regulatory stringency is lowest at the beginning of process development efforts. It increases as clinical trials progress into later stages, reaching its highest level during commercial manufacturing operations. So process development offers the best opportunities for a team to explore its control space and develop the scientific support and experience necessary to maximize the predictability and quality of the manufacturing process. Once commercial operations begin, such opportunities are much more limited.
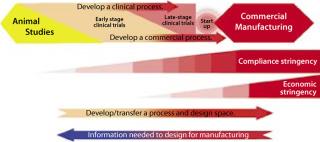
The better the understanding of a process before commercial start-up, the shorter will be the technology transfer time between process development and manufacturing teams, and thus the shorter the time to commercial release and return on investment. With opportunity costs for new drugs averaging over US$1.4 million/day, shortening tech transfer time offers significant commercial payback (2). Long after formal transfer is complete, it is the process development team that gets called in to troubleshoot a manufacturing process, make improvements, and deal with upsets.
Throughout process development and manufacturing work, the flow of vital information is bidirectional between process development and manufacturing groups. This is critical to the remaining elements of QbD.
Design-Space Development Relies on ScienceProcess development ultimately defines and gains approval of a manufacturing control space within the universe of possibilities about a process called a knowledge space. An approved manufacturing process can be operated within its control space to produce material that meets the required specifications regarding identity, potency, quality, and so on.
As a product moves through its life cycle, scale-up, economic, and other factors can require changes in the control scheme for that process, necessitating an updated control space. The scientific basis for a new control space is usually developed to cope with process shortcomings that arise long after the original process development work was done. Creating one can be a costly and inefficient process because it can trigger the need for new clinical studies.
With the publication of ICH Topic Q8, manufacturers can now develop an approvable design space in advance of commercial launch that anticipates and accommodates more than one control space (3). This allows them to make changes that move a process from one control space to another without the need for additional regulatory approval — provided that both control spaces remain within the approved design space.
To realize the benefits of such an approach, process development and manufacturing teams must document in the chemistry manufacturing and controls (CMC) section of a regulatory submission the scientific basis for approval of the design space for their process. Information comes partially from appropriately designed experiments that define and test the outer limits of the intended design space. This helps a manufacturer understand the effects on critical quality attributes as well as to define and characterize new critical process parameters that might arise in a new control space. At the time of process start-up and technology transfer, all supporting data and information must be readily available to the entire team.
Design for Manufacturability Uses Process InformationDeveloping and gaining approval for a control space and/or design space depends on making full use of prior knowledge and experience of the process development and manufacturing teams. Data concerning how previous processes behaved when subjected to the constraints of full-scale commercial operations provides a vital source of guidance for designing later processes to operate successfully within similar constraints.
The FDA uses the phrase design for manufacturability to describe the use in process development of information from prior manufacturing to consistently achieve an acceptable standard of predictability and quality. This type of information comes from accessing and analyzing actual data from prior manufacturing processes operating under the same or similar conditions as a process currently in development. This work identifies, correlates, and monitors the relationships between CPPs and CQAs under the well controlled, full-scale operating conditio
ns used for commercial manufacturing.
Other sources of valuable information used in design for manufacturing include data about the relationships between CPPs and CQAs collected during manufacturing process upsets (out-of-specification or OOS results). Such upsets are the practical equivalent of full-scale experiments that can also reveal previously unrecognized CPPs, control of which also must be built into the next process. Failure to use these valuable sources of information is generally due to
-
data not collected and/or not made available for analysis
-
CPP information too deeply buried in oceans of in-house data, or
-
a culture of collaboration and continuous process improvement is absent or lacks the proper supporting technologies.
QbD is also one of the most important efforts a team can undertake to bring about process improvements in an existing manufacturing process. The goal is to design improvements through increased process understanding — reducing process variability and moving towards real-time quality assurance. Figure 2 shows the major areas of focus in a process improvement cycle. Beginning in the lower left and moving clockwise, the following are used to routinely review the variability of CQAs:
-
on-demand access to all discrete and continuous manufacturing data (from their different data sources)
-
automated reporting and process trending
-
control chart and trending capabilities.
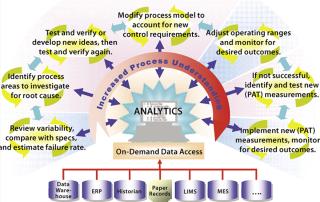
Visual and quantitative comparisons are made to the specifications, and process capability analyses are performed to determine the expected failure rates if an observed variability is left unaddressed. If the level of CQA variability is unacceptable, investigational analysis is undertaken to determine cause(s) and identify improvements that can be made to the process control strategy to correct the variability. An improved control strategy will either apply to the existing control space or necessitate a new control space (optimally still within the approved design space). This investigation can be as simple as reviewing a process in light of commonly known principles of design and control (sometimes called first principles), or it may require a detailed search for less obvious data correlations from previous process development and/or manufacturing work.
The iterative work of investigating the most likely causes of CQA variability relies heavily on the ability of individual team members to freely contribute ideas and test, reject, accept, and develop them easily in collaboration with other team members. First ideas are often rejected after initial testing, and they require further development and testing using additional data until root causes are found and verified using sound scientific methods.
So on-demand user access to data from multiple disparate sources (including paper records) is a practical necessity. If it takes weeks to obtain the information necessary to test each idea, then creativity and collaboration can dry up quickly, leaving problems unaddressed and unresolved for long periods. Analytical tools should be available immediately in the same environment as the retrieved data.
When the most likely causes of CQA variability have been identified, a modified process control strategy using existing process instrumentation can be tested with predefined success criteria under a planned deviation or interim operating procedure. Its results must verify that the control strategy has brought about the desired process improvement.
Implemented process control strategy adjustments may not bring about the desired improvement in CQA variability. Investigational work might provide no scientific basis for changing a control strategy based on existing process measurements. In either case, knowledge of first principles together with insights gained during the investigational analysis will most likely indicate both the type of new measurements needed for improved control and the process area requiring improvement.
New process measurements may require instruments that provide direct real-time measurements of the CQAs with unacceptable variability. Such on-line data would allow progress toward real-time quality assurance consistent with the goals of process analytical technology (PAT). Instruments must be tested in well-designed trials to assure that they produce the desired reduction in CQA variability. Control charting and other types of data analysis can show whether a process is then under sufficient control.
The individual process improvement tasks in Figure 2 can be repeated as indicated by the pairs of circular arrows around them. Regular ongoing tasks, for example, usually include process monitoring, trending, comparison with specifications, and process capability analyses. A team can move back and forth as needed between major tasks in the process improvement cycle. The goal is to progress steadily clockwise through the cycle, repeating as necessary, to achieve process understanding for sufficient control of CQA variability and keep the control space within an approved design space.
Progression through the process improvement cycle increases process understanding that a team can apply to existing processes. Consistent with the principles of design for manufacturing, such understanding also should be used in the design of the next new process to prevent recurrence of the same sources of process variability.
Collaboration and Data Access Are KeyIf process trending shows that the variability of CQAs is sufficiently controlled, the process improvement cycle need not be repeated. However, even when significant resources have been devoted over long periods to achieve sufficient process understanding during development of a new process or during full-scale operations, real-world experience has shown that process upsets will inevitably occur. They are an indication that a previously unidentified CPP has surfaced, and the cycle of process improvement must be repeated as necessary to bring a process back under control. A collaborative environment with on-demand data access for end users — integrated with analysis and reporting capabilities that span geographic locations — will help get a process through such upsets in the shortest possible time. Ideally, the frequency of opportunities presented by process upsets decreases over time until real-time quality assurance can be achieved — hopefully before the end of the useful commercial lifetime of a drug.
Excellence Achieved Through QbDWhen fully implemented, QbD means that all critical sources of process variability have been identified, measured, and understood so that they can be controlled by the manufacturing process itself. The resulting business benefits are significant: reduced batch-failure rates, reduced final-product testing, lower batch-release costs, faster regulatory approval of new product applications and process changes, fewer inspections of manufacturing sites, and a more predictable supply of product. Such benefits translate into significant reductions in working capital requirements, resource costs, and time to value. And those bottom-line gains ultimately pave the way
for additional top-line growth.