Discovery, development, and commercialization of novel biologics frequently involve collaboration between two or more companies. In the context of these business relationships, transfer of technology from one institution to another is a crucial step that needs to be executed flawlessly and rapidly. Follow-up activities usually include the development of productive, reliable, and scalable processes and are equally important because they are usually on the critical path to market.
PRODUCT FOCUS: MONOCLONAL ANTIBODIES
PROCESS FOCUS: TECHNOLOGY TRANSFER (PRODUCTION) AND ANALYTICAL METHODS DEVELOPMENT
WHO SHOULD READ: PRODUCTION AND PROCESS DEVELOPMENT, MANUFACTURING
KEYWORDS: IGF-1R, EXPRESSION, TECH TRANSFER, CELL-LINE CULTIVATION, SIZE-EXCLUSION CHROMATOGRAPHY, ISOELECTRIC FOCUSING, MATRIX-ASSISTED LASER DESORPTION/IONIZATION TIME-OF-FLIGHT MASS SPECTROMETRY, ELECTROSPRAY IONIZATION MASS SPECTROMETRY
LEVEL: INTERMEDIATE
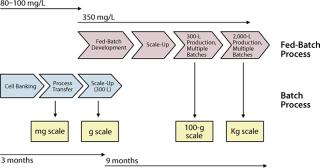
In the context of the licensing of a therapeutic antibody for the treatment of cancers over-expressing the insulin-like growth factor I receptor (IGF-1R), this two-part article describes the steps and timelines that led to successful and rapid transfer of technology and its subsequent scale-up. These activities produced antibody used in preclinical and early clinical evaluations. Transfer of technology and its scale-up by ∼150-fold was completed within a period of three months, and development of a more productive process and its ∼1,000-fold scale-up was completed within about nine months. In the March 2008 issue of BioProcess International, the first half of this article listed materials and methods for the studies, detailed assay development and analytical methods involved, and provided analytical results before summarizing the initial scale-up efforts involving a batch process. This month concludes with a detailed discussion of the fed-batch process scale-up.
Fed-batch Process DevelopmentIn parallel with scaling up the batch process described in Part One, a fed-batch process was rapidly developed. We quickly evaluated a baseline feed solution previously used for another NS0 culture process for its potential to increase process productivity here. Then we further refined process performance through implementing iterated modifications to the composition of the feeding solution and fine-tune the process to the specific cells used. These experiments were performed at the shake-flask scale, a previously proven model (1).

LUC ASBURY (WWW.SXC.HU)
Figure 4 compares performance of the batch process to that of a preexisting fed-batch process. Panels A and B compare growth, viability, and antibody production to show that implementation of the platform fed-batch process increased maximum cell concentration by 50% while dramatically reducing cell death rates. This improvement in cell cultivation correlates with a 2.30-fold increase in final antibody titer (from 100 mg/L to 230 mg/L).

Panel C shows that whereas during batch cultivation glucose is consumed to but half of its initial concentration, during fed-batch cultivation glucose is almost entirely depleted by the seventh day. Clearly in fed-batch cultivation glucose becomes limiting for the specific clone cultivated, which necessitates optimizing the availability of this nutrient. The same panel shows that lactate transiently accumulated after four days of cultivation and was entirely consumed by the end of the fed-batch cycle.
Because of cells overexpressing glutamine synthase, no ammonia accumulation was observed under either batch or fed-batch cultivations (data not shown). The feeding solution we used was formulated to minimize necessary addition of salts and buffers. So osmolality of the fed-batch culture was maintained around 300 mOsm/kg, a value similar to that achieved in batch cultivation and well within the range of tolerability for NS0 cells (2).
Feed-Solution Optimization: Following those encouraging results, we modified the feeding solution to improve performance, adapt it to the specific clone, reduce cost, and eventually improve the ease of formulation. The first feed modification removed recombinant human insulin (rHI), an expensive and difficult-to-procure insulin commonly used as a growth factor in cell culture media. It has been reported that zinc can be used as a replacement for insulin (3). So we evaluated ZnCl2 as a replacement for rHI. Table 3 shows that replacing rHI with ZnCl2 (feed modification 1) supported similar cell growth, but also slightly reduced antibody production (from 243 mg/L to 218 mg/L). Under these conditions a higher rate of cell death was observed, as indicated by the low viability at the end of the cultivation cycle. Interestingly, both feeds demonstrated similar lactate and glucose trends (data not shown).
Table 3: Fed-batch process performance of various feed solutions; feed modification 1 = insulin replaced with ZnCL2, feed modification 2 = cholesterol concentration increased, and feed modification 3 = addition of hydrolysate

The second modification made to the feed solution was to increase cholesterol concentration. NS0 cells are cholesterol auxotrophs, which require the addition of an exogenous supply of cholesterol to survive (4). In addition to the CLC medium (a mixture of cholesterol, fatty acids, and cyclodextrin from Invitrogen, www.invitrogen.com), we investigated the additional supplementation of LS1000 (from HyClone, www.hyclone.com), a solution made up of cholesterol and cyclodextrin (feed modification 2). This increased the maximum cell concentration from 2.8 × 106 to 3.2 × 106 vc/ml. This further
correlated with a MAb titer increase from 218 mg/L to 290 mg/L. Increasing the amount of cholesterol also appeared to improve cell metabolism. Although the glucose consumption pattern was unaffected, the maximum lactate accumulation decreased about 50% (data not shown). In addition, final viability also improved (Table 3).
The third modification we made to the composition of the feed solution was addition of a hydrolysate. Use of hydrolysates can significantly improve cell viability, growth, and correlatively the yield of MAb (5,6,7,8,9,10). After screening several at different concentrations, we found that the use of yeast hydrolysate at 10 g/L of feed solution was best for this specific process. Table 3 shows that this addition (feed modification 3) gave a 30% increase in maximum cell concentration and boosted final antibody concentration to ∼400 mg/L. Lactate profiles mimicked those of feed modification 2, but a corresponding increase in glucose consumption was probably due to the increased cell population (data not shown). Daily osmolality measurements showed that it stayed below 350 mOsm/kg at all times and was not deemed to have the potential to negatively affect the health of the cells (2).
Demonstrating the Process at Laboratory ScaleFollowing completion of our small-scale fed-batch process development studies, we implemented the process in bioreactors at 3-L, 30-L, and 300-L scales to evaluate scalability of the newly developed fed-batch process. To simplify the operation and reduce volume gains, certain modifications were made: The number of feed-solution additions was reduced from five to three, using solutions that were more concentrated, and the delivery of cholesterol by LS1000 alone was implemented. Process performance equivalency of the resulting feeding regimen was demonstrated at the shake-flask and 3-L scales (data not shown).
Implementation of the fed-batch process was then first demonstrated at the 3-L scale and then rapidly scaled up to 30-L and 300-L bioreactors. Figure 5 compares growth, viability, antibody production, glucose, and lactate kinetics. Table 4 summarizes the process performance data for a representative culture at each scale. Similar performance was achieved at each scale to yield about 350 mg/L of MAb. Table 4 also shows that good reproducibility was achieved within the 3-L and 300-L scale cultivations, as supported by the tight standard deviations. Taken together, these data demonstrated process robustness and proof of scale-up in laboratory bioreactors over a 150-fold increase in scale.

Table 4: Fed-batch performance at various laboratory scales

Scale-up in large facilities presents multiple challenges, both in terms of operation and process parameter scale-up. For this specific process, two examples outline how these kinds of challenges can be addressed: feed formulation and bioreactor scale-up.
Feed Formulation and Manufacturing: Early in development of our platform feed solution, the feed was prepared in-house from several separate solution components as a somewhat dilute solution. To minimize broth dilution later in development, the feed became an increasingly concentrated solution whose manufacture was eventually outsourced and scaled up. After several attempts, we found that manufacture of our feed solution was not fully scalable to large scales, which involved formation of a precipitate. To yield a similar feed solution without precipitation, we reengineered preparation of the feed solution by calculating the solubility of components in partially precipitated feed solution.
The revised formulation protocol used a higher pH to prevent precipitation during manufacture and cold storage of the feed solution. This revised feed solution was slightly more dilute than the original formulation because of the pH adjustment step, so a final titration experiment was completed to ensure process and product equivalency. Table 5 shows that cell growth and antibody production performance were comparable for cultures using both revised and original feed formulations. SDS-PAGE analyses (reduced and nonreduced) further demonstrated equivalent product profiles.
Table 5: Performance comparability of reengineered feed solution

Bioreactor Scale-Up Strategy: Development and scale-up of the fed-batch culture process was specifically targeted to achieve process fit at the intended pilot plant, where stirred-tank capacity was available at the 2,000-L scale. Because one primary consideration in this strategy was to prevent the need for an antifoam agent, minimizing foam formation was of some importance.
Table 6 presents vessel geometry and relevant agitation and aeration parameters for the 300-L and 2,000-L bioreactors. The scale-up approach used geometric similarity of vessels with agitation operated at constant power input per unit volume across scales. Because volumetric mass transfer (kLa) is primarily a function of power input per unit volume and superficial gas velocity (11,12,13), we based our chosen agitation speed on constant specific power input to maintain similar oxygen mass transfer capacity between scales. As expected, foam generation was primarily a function of superficial velocity. However, surface foam accumulation was also found to be enhanced by agitation power input. Therefore, the specific power input was kept low at about 10 W/m3.
Table 6: Vessel geometry and relevant agitation and aeration pa
rameters for the 300-L and 2,000-L bioreactors.

The overall flow rate of the three gases we used was scaled separately with respect to minimum and maximum sparge gas flow while the combined minimum sparge rate was scaled on constant VVM and the combined maximum sparge rate was scaled on constant superficial velocity (vs). We designed the maximum overall sparge gas flow rate for dissolved oxygen control to stay well under a vs of 0.070 cm/s, below which there was little to no foam generation. Although the sparge gas flow rate was kept at a relatively low level, it was sufficient to keep the partial pressure of CO2 to 100 mmHg, a level acceptable for cultivating NS0 cells (14). Table 6 lists some operating parameters that were selected for each scale.
Process Performance at 2,000-L ScaleOnce all issues pertaining to process scale were solved, we executed our first batch at the 2,000-L. Figure 6 shows that when compared with a typical 300-L fed batch (Figure 5), the 2,000-L fed batch performed very closely in cell growth, viability, and antibody production. Table 7 clearly shows that process performance was very consistent across six independent 2,000-L fed batches. When considered together, these data show that both scale-up and process robustness were achieved at the large pilot scale. Figure 6 shows excellent reproducibility for growth (6A), product titer (6B), metabolic profiles (6C), and viability (6D) across six different 2,000-L cultivations.
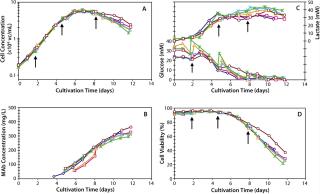
Table 7: Fed-batch performance at pilot-plant scale

Table 7: Fe
Following separation of the cells, the monoclonal antibody (MAb) they produced was purified by several chromatographic steps, including protein A. The purified product served to demonstrate the consistency of product quality, as evaluated by standard biochemical assays such as simple protein integrity by sodium-dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE) to charge variants by isoelectric focusing (IEF) and glycan distribution. Overall, product consistency was maintained throughout scale-up and across all batches performed at the 2,000-L scale. Full description of this purification process and its performance will be the subject of a future paper.
Project SuccessWe have clearly outlined the various steps that led to successful transfer of a cell cultivation process from Pierre Fabre Medicament to Merck and Co. Despite moderate productivity (∼10 pg/cell/day), we decided to use the original GS-NS0 clone to support an aggressive timeline toward the initiation of preclinical and clinical evaluations.
After completion and testing of the cell banks, rapid implementation of our process was demonstrated in laboratory bioreactors for production of analytically comparable MAb. This phase is crucial because it establishes internal know-how and familiarity with a specific project. To achieve maximum efficiency during that early phase of technology transfer, we implemented and demonstrated equivalent a concurrent minor process change, the elimination of a cell concentration step at the time of bioreactor inoculation. These changes greatly facilitated rapid 300-fold scale-up of the modified process. This first scale-up was completed within three months of the arrival of the cells at Merck and supported the production of tens of grams of MAb that were critical in supporting purification, formulation, and stability studies as well as development of various analytical methods.
Concurrent with the implementation and scale up of the batch process, a fed-batch process adapted from a preexisting platform was rapidly developed and scaled up in laboratory bioreactors. Implementation of fed-batch technology is a well-established method for increasing cell density of a culture while extending cell viability (15, 16). But the approach usually requires specific adaptation of a general protocol to each individual clone because cells transfected with rDNA and overexpressing a recombinant antibody tend to behave uniquely. Following a few iterations in the adaptation of the fed-batch protocol, we increased process performance by 3.5-fold over the batch process. More important was the successful scale-up to the 300-L scale, which supported the production of large amounts of MAb for preclinical evaluations. This part of the process enabled the product development program to be rapidly supplied with several hundred of grams of MAb.
Implementation at pilot-plant scale (2,000 L) first required large-scale production of a homogeneous feed solution that did not exhibit any precipitation in storage. Because they contain highly concentrated ingredients such as amino acids and vitamins, feed solutions tend to exhibit poor solubility, so their development is an important step in implementing a successful fed-batch process. The feed solution we developed and industrialized proved to be highly consistent in supporting process performance.
Scaling up mammalian cell processes also requires attention to aeration and mixing conditions. Balance must be achieved between providing sufficient oxygen along with stripping CO2 from a culture and elevated gas sparge rates that can cause foaming and/or cell damage (12, 13). Calculated values for scale-up of sparging and mixing proved to be effective, and the translation of process performance was excellent upon scale-up to a 2,000-L bioreactor, with cell production levels and cell viability profiles very comparable across all scales. Testimony to the robustness of our process is the <10% deviation in MAb production across six 2,000-L successive cultivations in the pilot plant.
Taken together, the data presented here and in Part One show that a process transfer completed between two independent organizations can be successful if it is managed closely in the technical arena, which includes close communicat
ion between all scientists and engineers involved. This approach allowed our team to keep the time of completion for all activities including preparation of clinical supplies under one year (Figure 7). And this permitted rapid evaluation of the antibody in both preclinical and clinical settings and helped position the overall program for success.