The level or intensity of product and process understanding that can or should be achieved beyond the acceptable minimum level promises to be the scope of a continuing debate among biotech industry and its regulators. In practice, the path of increased understanding may follow a series of incremental steps toward the desired state (Figure 1) after a product launch. Realistically that is expected to occur when the level of product and process understanding has reached or slightly exceeded the minimum regulatory acceptable level. Development of such understanding beyond information collected from product and process characterization studies (see the appendices online) during development can come from using a process analytical technology (PAT) approach for process monitoring or using newer scientific approaches such as systems biology.
Continual or Continuous Improvement
Some authors prefer the term continual improvement over continuous improvement. Although both are similar, they may invoke different meanings within a certain context. We chose to use the first term throughout these chapters for consistency.


WWW.PHOTOS.COM
Process Analytical Chemistry: Under the PAT paradigm, real-time (RT) assessment of critical quality attributes (CQAs) either directly or inferred (surrogate) through measurements of critical process parameters (CPPs) comes from process measurements, preferably conducted in-line or on-line. If CQAs are merely inferred through process measurements, then it’s essential to know how derived such measurements are from the respective quality attributes they portray. One precursor to implementation of advanced feed-back process control strategies (see below) is the need for in-line or on-line process measurements. Unlike in many other process-based industries, for the biopharmaceutical industry such measurements have been principally hampered by
-
limited availability of suitable sensors or a lack of suitable analytical measurement technology altogether, including system components for retrieval of process samples under sterile conditions and system connectivity
-
challenges surrounding sensor calibration, multivariate data analysis algorithms, and analytical method validation (compared with off-line univariate assay validation)
-
incomplete understanding of the relationships between process parameters and affected product quality attributes, particularly under dynamic process conditions.
Among the many considerations to be made regarding implementation of process analytical chemistry (PAC) analyzers for process monitoring is identifying those actually relevant to managing the continuous identification and prediction stages in a dynamic process. PAT is not about adding yet another process analyzer to the analytical tool box just because it is available.
One way to further RT process monitoring is to look at what analytical assays have been applied to date in a manufacturing process in the first place and “engineer” them in. Examples include potency/activity, identity, and purity assays in QC laboratories — either as in-process assays or as end-product release assays. Searching for appropriate monitoring technology may determine whether the related quality attributes actually can be assessed in real-time. Are technologies (sensors) even available to accomplish this? Of course, in determining the need for application of RT capabilities, you need to distinguish between assays used solely during R&D and those that are fully validated for routine use in manufacturing — not to mention compendial assays that are expected as part of a CMC regulatory filing.
Yet another approach is to consider each unit processing operation (UPO) and identify those that already incorporate many interactive controls (e.g., bioreactors in production and chromatography in downstream processing) as obvious candidates for PAT approaches. Searching for current or additional process parameters that provide markers for in-process product quality attributes may eventually render the need for certain bioassays or procedural operating steps obsolete. For example, determination of the proper time to harvest from a bioreactor culture may come from analyzing specific metabolic indicators to reveal CQAs, rather than merely relying on some previously identified fixed time point.
The Systems Biology Approach: Moving beyond traditional “reductionist” biological science approaches, systems biology (SB) brings the biological, analytical, information management, and engineering disciplines into a holistic approach to a mechanistic characterization of biological systems (1). Not surprising, this is considered by some as the avant-garde of science and technology: to reveal the actual systems and molecular basis of biological forms, function, and behavior.
SB provides a means to study the complex interactions of biological networks and their components and reveal how they determine overall function. It promises unprecedented levels of understanding for paving the way to dramatically improve our understanding in such topics as cell biology and human health. The approach was actually conceived in the 1950s, but only recently did four fundamental areas advance sufficiently to begin a practical application of the concept.
First, developments in such fields as genomics, proteomics, and high-content screening are providing detailed biochemistries to feed SB. Second, new technological platforms such as yeast-two-hybrid screens and RT-PCR arrays are providing new tools for molecular probing. Third, advances in analytics (including new hybrid and tandem technologies and platforms providing automated, robotic, high-throughput, and multiplexed analytics) support comprehensive data acquisition. And fourth, advances in bioinformatics, computational biology, systems and modeling theory, and information technology provide the power to integrate and manipulate the enormous volume of data generated.
Biopharmacology presents a number of distinct applications for SB, such as comprehensive and specific descriptions of the molecular mode of action of drugs; the organelle and cellular processes involved in biosynthesis of large, complex biomolecules; and the determinants of cellular response to changes in process conditions. The SB approach supports development of a holistic view of cell-based manufacturing processes by increasing and synergizing knowledge across multiple science and engineering disciplines.
Bioproduction As an Example: Increasing our understanding of the dynamic, multifactorial relationship between cell function and relevant environmental and material characteristics, for example, leads to a more mechanistic understa
nding of the entire production process. By providing a comprehensive, quantitative, and predictive knowledge of a cell’s behavior in application, SB tempts us with the possibility of revealing true process dynamics. In particular, it offers a unique opportunity to elucidate underlying interactive cellular mechanisms that determine optimal process conditions and then strictly correlate in-process material characteristics and process parameters to CQAs.
One example of such application pertains to the dynamic mechanics and cellular process determinants of a protein population in bioproduction. SB provides a departure from current approaches involving many standalone analytical techniques to measure static product attributes in local and unidimensional pathways. One limitation of conventional approaches comes in our attempt to describe the complexity of an entire glycosylated protein or even the quality status of its manufacturing process as an ensemble of average states of even indirect or surrogate characteristics. Averages do not capture the contributions and actual distributions of individual parameters and actually can be deceiving when describing biological products because of inherent microheterogeneities. Even accurate and descriptive measurements revealed at one time-point and after the fact are useless for such activities as process control.
Combining SB results with multiparametric and high-content assays of either production cell metabolism or product characteristics, then relating them quantitatively and in real-time to CPPs, should provide for better understanding of dynamic cellular responses and product profiles in the changing environment of a bioreactor. In fact, we believe that certain QbD goals in some platforms won’t be feasible without SB. It may be that for complex glycosylated products made by mammalian cell culture, development of a comprehensive design space or accomplishment of real-time release (RTR) may be impractical without the understanding provided by SB. So it not only finds an immediate usefulness in biopharmaceutical discovery programs, but also represents a new and truly unique resource in the design and control of cell-based manufacturing processes.
In any event, results from SB approaches to biopharmaceutical applications should also feed into advanced process modeling activities, data mining, and formal experimental design across a design space. The growing appreciation of SB as a promising field in biotechnology is evidenced by the fact that CASHE’s Foundations of Systems Biology in Engineering (FOSBE) is now preparing for its third annual conference (www.ist.uni-stuttgart.de/fosbe).
No single company or university can hope to fully develop this new approach on its own. To efficiently work in a larger context using automated annotation and Web-based, large-scale data integration and mining recourses, it will be imperative to standardize such parameters as experimental conditions, analysis metrics, and reporting formats following best industry practices. This would enable data capture to be meaningful for model development and process understanding across the industry.
Enhancing Process ControlConsidering the principles of the quality-by-design (QbD) philosophy, designing a robust process (Figure 2, and see the appendix online) depends on the interplay of two distinct factors: the level of process understanding achieved and the level of process control implemented.
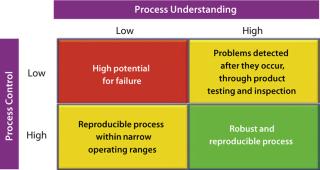
It is important to recognize that even with a low level of process understanding, a reproducible manufacturing process can be achieved if it involves a high level of process control. Defining process robustness as the ability of a process to tolerate variability in materials and process/equipment changes without negative affects on quality makes it apparent that a reproducible process does not necessarily equate to a robust one. Processes that are both reproducible and robust can be achieved only with high levels of process understanding and process control. So it is not surprising that the PAT guidance emphasizes both factors.
For example, a PAT approach may be implemented through specific and timely in-process (e.g., at-line, on-line) multivariate measurements of actual in-process material parameters and various critical-to-product-quality parameters/attributes with a process feed-back loop (Figure 3). This represents a significant departure from traditional manufacturing processes, in which such parameters/attributes are assessed to a lesser degree, at a different time, and often at a different location (the QC laboratory). They rely primarily on process equipment or SOPs to determine univariate physical process parameter set-points to support robustness and end-product quality.
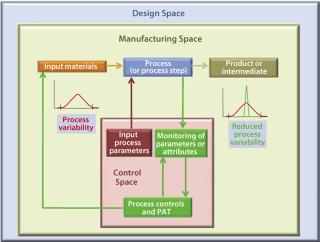
A PAT-based process control implementation requires
-
dedicated PAC tools or process analyzers (implying suitable sensor technology, preferably in on-line settings, that captures process data hopefully in real-time)
-
software tools to analyze incoming multivariate process data (to extract their individual, discriminative, and contributing measurements)
-
specific control algorithms that combine deconvoluted, multivariate data (often with more common, simple, single-parameter physical measurements).
That constitutes a basis for advanced process control strategies (e.g., model predictive control) ensuring that both a desired process trajectory and end-product quality are achieved through in-process feedback (closed-loop control). The underlying control principle is to apply knowledge gained from higher process understanding to mechanistically link critical product attributes (CPAs) with measurements of process variables supporting the institution of optimal process control.
Process Validation As Continuous Process VerificationProcess validation is necessary to an overall control strategy both for assuring product quality and fulfilling regulatory requirements. One of its hallmarks is the successful completion of typically three consecutive batches (2, 3), referred to as conformance lots, that are manufactured under routine commercial operating conditions. They are intended to measure inherent reproducibility and establish that process variation falls within the
preestablished specifications. However, many experts have pointed out that the mere ability to manufacture just three conformance lots within one time period does not necessarily demonstrate process capability. Under the new operating paradigm, companies can conceivably validate each batch through continuous process verification — while it is being produced — which may even eventually replace the need to manufacture conformance lots. Notably, a new compliance policy guide contains no specific reference to three conformance lots (4). Although recombinant protein drug products are not specifically addressed, the guide does in general recognize the role of emerging advanced-engineering principles and control technologies in ensuring batch quality.
The 1987 process validation guidance is currently under revision to incorporate the modern concepts of QbD and PAT (3). It promises to focus on a validation life-cycle (from product and process development to commercial retirement) and to emphasize elements of process design, understanding, and capability; continual improvement; risk management; and change control (5). Although under the new operating paradigm process validation is intended to confirm both the design space and the applied control strategy, challenges remain on how best to implement it. For example, comprehensive process variation — and consequently, process robustness and capability — cannot realistically be tested at the commercial scale. Instead, adequate process characterization (see the appendix online) must ensure that a process is inherently robust by design because, even if feasible, it is inappropriate to learn about previously unknown aspects of a process during the manufacture of conformance lots.
Fortunately, industry experts are involved in the unfolding debate and are proposing pragmatic solutions based on integrated systems validation plan development. Such a plan provides a simplified structure for describing a process, assessing its variability, identifying CPPs, and setting up a system to maintain that process in a state of control (6) — all while following the intent of QbD and PAT for applying novel technologies where and when appropriate.
In the new framework, manufacturers of biopharmaceuticals now can evaluate and propose alternative approaches for validation and qualification to transition from the traditional “test and document everything” toward a life-cycle approach focusing on what is critical for product quality. One example is a modular validation approach that uses data from validation or characterization studies performed on a specific UPO for one protein in support of validating the manufacturing process for a related protein. Consensus standards — such as those currently being developed by ASTM as described in the Technical Committee E55 box — should facilitate this process.
Toward Integrated Knowledge ManagementIn general, the biopharmaceutical industry is not reaping the maximum value of data it collects, whether for future process design, current process optimization, identifying process improvement opportunities, or even proactive troubleshooting. All too often, data are collected merely for regulatory compliance — in data “islands” with limited structure, accessibility, and analysis capabilities beyond their immediate purpose.
An often inconvenient truth is becoming apparent regarding the means of acquiring sufficient product knowledge and process understanding to provide a desired measure of effective process design and control activities. Such an ability apparently depends on two quite formidable primary activities: integration of unique, complementary, and often large data sets from multiple control and analytical platforms across an entire manufacturing space; and extending that activity to product and process characterization (see the appendix online) across an entire design space.
For example, PAT-enabled manufacturing processes often measure additional process/product variables. They often generate several orders of magnitude more data than processes that rely solely on laboratory testing. Such data originate from a number of disparate analytical technologies for on-line process monitoring and control, fault detection/diagnosis, and product quality prediction in real-time — extending across an array of process control platforms. For data integration, information-generating processes must be connected to a higher-level process — e.g., as part of an enterprise manufacturing system (EMS) or manufacturing execution system (MES) with electronic batch record (EBR) and advanced process control capabilities. In fact, ASTM subcommittee E55.03 is currently developing a standard practice for PAT data management, which is highly anticipated by information technology developers.
Achieving complete data integration over an entire product life-cycle requires yet another, higher level of knowledge management architecture. This can be accomplished by means of an enterprise document management system (EDMS) to ensure project-wide data availability, with adequate statistical and graphical tools for data reconciliation, mining, and analysis. Such systems should include modeling capabilities to reveal hidden relationships between process and product measurements and link key performance indicators (KPIs) and key quality indicators (KQIs) with their underlying design elements, thus identifying opportunities for continual improvement.
The concept of data integration is far from trivial. One encouraging fact is that acquiring and correlating all those data points into a usable design and control knowledge database is now much more attainable than ever before using the powerful informatics platforms and data processing software now available. One advantage of modern informatics tools is that they are based on open-system architectures, unlike many of the legacy IT systems that still persist today. Nevertheless, special attention needs to be devoted to integrating individual subsystems across the functional boundaries of an entire organization because each subsystem usually has its own goals and constraints that must be satisfied in addition to those on a higher system level.
One key to optimizing biopharmaceutical manufacturing processes under the QbD paradigm will undoubtedly be to make product and process related data more visible in the context in which it applies. For example, in process design this would bolster the development of new design and control approaches. In manufacturing, it could allow for seamless integration of PAC tools or process analyzers to effect model predictive control (MPC). Such “visibility” will be important not only across all operational activities at the company level, but also for regulatory authorities as part of approval, inspection, and monitoring processes. And data integration solutions must meet 21 CFR Part 11 (7) requirements with both manual and technical procedural controls designed to ensure the authenticity, integrity, and any required confidentiality of electronic records.
ASTM TECHNICAL COMMITTEE E55
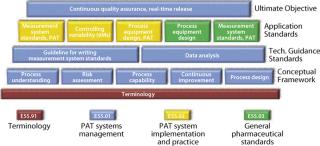
ASTM is developing standards for process design, control, performance, and quality through technical committee E55 on Pharmaceutical Application of Analytical Technology, which has been broadened and renamed as Technical Committee E55 on Manufacture of Pharmaceutical Products (12). E55 is active in such areas as risk assessment and mitigation, PAT, process sampling and capability, data management, continuous quality verification (CQV), and systems qualification (Figure 5). ASTM has a long histo
ry in the development of standards by taking a transparent and balanced approach that allows representatives from all sectors with an interest in the use of a standard to develop consensus. Participation of the FDA in the development process does not necessary imply adoption; however, the agency generally encourages use of these consensus standards where practical and can cite them in guidance documents and regulation.
For a complete overview and most current information visit www.astm.org/commit/subcommit/E55.htm. Here are some selected standards under the jurisdiction of ASTM technical committee E55.
Subcommittee E55.01 on PAT System Management
WK5930: Standard Practice for Risk Management As It Impacts the Design and Development of Processes for Pharmaceutical Manufacture (under development)
WK5935: Standard Practice for Process Understanding Related to Pharmaceutical Manufacturing and Control (under development)
WK9192: Standard Guide for the Application of Continuous Processing Technology to the Manufacture of Pharmaceutical Products (under development)
WK9645: Standard Guide for Application of Process Capability (under development)
WK12892: Standard Practice for Process Sampling (under development)
WK15151: Practice for Sampling Consideration for Pharmaceutical Applications of Process Analytical Technologies (under development)
Subcommittee E55.02 on PAT System Implementation and Practice
E2474-06: Standard Practice for Pharmaceutical Process Design Utilizing Process Analytical Technology (published November 2006)
WK4185: Test Method for the Measurement of Thermal Effusivity of Raw and Process Materials (under development)
WK5931: Standard Practice for PAT Management (under development)
WK9182: Standard Practice for Qualification of PAT Systems (under development)
WK9191: Standard Practices for Multivariate Analysis Related to Process Analytical Technology (under development)
WK13538: Standard Practice for Identification of Critical Attributes of Raw Materials in Pharmaceutical Industry (under development)
Subcommittee E55.03 on General Pharmaceutical Standards
E2500-07: Standard Guide for Specification, Design, and Verification of Pharmaceutical and Biopharmaceutical Manufacturing Systems and Equipment (published August 2007)
WK9864: Standard Guide to a Science and Risk-Based Approach to Qualification of Biopharmaceutical and Pharmaceutical Manufacturing Systems (under development)
WK9935: Standard Guide for the Application of Continuous Quality Verification to Pharmaceutical Manufacturing (under development)
WK11898: On-Line Total Organic Carbon (TOC) Method Validation in Pharmaceutical Waters (under development)
WK15778: Guide for Science-Based and Risk-Based Cleaning Process Development and Validation (under development)
WK16888: Guide for Validation of PAT Methods (under development)
Subcommittee E55.91 on Terminology
E2363-06a: Standard Terminology Relating to Process Analytical Technology in the Pharmaceutical Industry (published July 2006, currently under revision)
WK4187: Standard Terminology Relating to Process Analytical Technology in the Pharmaceutical Industry (under development)
Figure 4 presents a conceptual design and control model for establishing a biopharmaceutical manufacturing process based on the QbD philosophy with an integrated, process-centric systems perspective. It depicts an implementation path using risk-based, continual improvement, and PAT principles. Where appropriate, it directs a distinct SB approach toward process understanding. The model should apply equally well for designing new processes and for optimizing existing ones. In the current harmonized framework, PAT and SB are two essential enabling technologies.
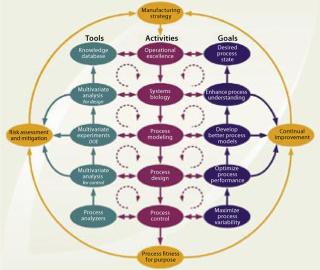
The principal goal of process design and control under the new operating paradigm is to provide an efficient and well-understood manufacturing process in which all sources of variation are defined and the critical ones are controlled. Specifically, product quality attributes should be accurately and reliably predicted over an applied process control space, and all sources of nonexceptional variation should be “directly” managed by the process itself. This condition, in which end-product variation is at an acceptable (minimal) level, is referred to as the desired state of a process. This implies that CPAs are controlled to their respective target levels for all individual product units manufactured.
A manufacturing process that is fit for its purpose must produce a uniform product. The presence of common-cause variability necessitates that the dispersion of quality values for finished units in a batch be such that, in the absence of special-cause variability, they can be characterized by a symmetrical baseline range within acceptable lower and upper specification limits. These processes must consistently supply the stipulated quality from unit to unit and batch to batch throughout a product’s life-cycle.
Generally, the capability of a process can be defined as the degree to which its predetermined specification range exceeds apparent variability. This gives rise to the quality metrics of process capability (CP) and process capability index (CPK) used by some to assess the validity of manufacturing processes (8). Although these can be excellent performance indicators, they are calculated under the assumption that a process is in statistical control and that related data follow a normal distribution. This may not necessarily be true, so they should be used with care.
A capable process can be achieved through development or optimization activities to minimize process variation by applying them in two overall frameworks: risk assessment and mitigation using formal risk-evaluation methods and continual improvement of process quality throughout a product life-cycle. Each of those frameworks is represented as an iterative loop in Figure 4.
Once a manufacturing strategy for
a particular product has been formalized, operational excellence (Op Ex) determines an overall process-centered implementation methodology. This is chosen by, for example, applying “lean” manufacturing (reducing the number of manufacturing process steps) and six sigma (enhancing the quality level of manufacturing process steps) principles that specifically focus on improving production efficiency, streamlining operations, and reducing process variability. Because an inherent objective of Op Ex is achieving process robustness, it correlates directly with establishing a desired process state. It also reflects application of current product knowledge and process understanding (the knowledge space). Independent of the actual implementation methodology applied, the proposed path of Op Ex includes consecutive design activities (each with its own subset goals, enabling tools, and deliverables) from such sources as SB, process modeling and design, process control, and ultimately validation to achieve a manufacturing process that is fit for its purpose. All activities along that path are purposely implemented to actively enhance process understanding and minimize variability.
The process design and control model in Figure 4 allows for flexibility in the execution of design and control activities. It does not imply that a proportionate amount of time must be allocated to each proposed step. The model further allows for iterations between (or beyond) given adjacent activities, as well as between their corresponding tools or goals, to allow for a phased progression of activities as required by each specific product or process. This model is our suggestion of one comprehensive and universal approach to mapping out an integrated and harmonized network of currently popular principles with their interrelated connections. It is merely intended as an aid in understanding the elements of the new initiatives. As such it may provide a general starting point to be abbreviated, extended, or otherwise modified as each specific application or interpretation dictates.
Implementation Approaches: Bottom-Up or Top-DownImplementing novel process design and control or improvement programs can follow one of two principal scenarios: bottom-up or top-down. These describe metaphorical directions indicating the interconnection of design and manufacturing space (Figure 1 in Chapter 2). In application, the labels are generalizations; many real-life programs use at least some elements of both approaches.
The Bottom-Up Approach starts in a development laboratory, where a product concept is implemented through product and process characterization studies (see the appendix online) concurrent with the initiation of process and control design activities to define a design space. These activities are conducted well before a technically feasible process can be implemented in the manufacturing space. This approach provides the best opportunity for meeting the highest unified goals of coordinated Op Ex, QbD, QRM, and PAT programs, and it could provide for true process innovation. Although bottom-up design is often the most demanding, it is nevertheless the most common choice in the development of new processes. It can even be successfully applied to the continual improvement of those that have been previously designed or implemented.
The Top-Down Approach begins at the opposite end of a development path: in a production area, where process design and control programs for a given manufacturing space are conceived that ultimately will be (somewhat “backwardly”) integrated into a design space. Typically a productivity improvement appraisal (PAI) is conducted to analyze an existing process (or steps thereof) and determine those that may benefit from a PAT-directed developmental approach (9).
That kind of PAI (not to be confused with another, the preapproval inspection by regulatory authorities) specifically focuses on analyzing critical operating data (COD); on benchmarking against best industry practices; and on determining overall equipment effectiveness (OEE), and key performance/quality indicators (KPIs/KQIs). Cost reduction is also a factor. Such process improvement programs are often designed to address process challenges that have been previously identified through data mining of existing batch records, reviewing out-of-speciation (OOS) reports, or corrective and preventive action (CAPA) investigations.
At first, the top-down approach may seem to be the most promising for achieving immediate gains. However, expectations for quick results may be thwarted if data analysis ultimately points to more fundamental process issues that are much more efficiently addressed directly in a design space (10, 11).
The actual approach chosen depends in large part on such tangible factors as the scope of an initial project; the stage, maturity, and robustness of the process at hand; allocated budget and resources (both human and otherwise); and available technologies and success metrics. However, it may equally depend on corporate philosophy, history, and culture, which of course differs greatly between biopharmaceutical manufacturers.