The only method of stable and long-term — practically infinite — preservation and storage of perishable biological materials (biostabilization) is to keep them in a glassy (vitreous) state. This was understood by Father Luyet when he titled his pioneering works in the 1930s, “The Vitrification of Organic Colloids and of Protoplasm” and “Revival of Frog's Spermatozoa Vitrified in Liquid Air” (1,2). He and other pioneers of the cryobiological frontiers clearly understood that only a glassy state would ensure stable and nonlethal preservation of cells.
Over time, a number of biopreservation methods were developed (3). Slow freezing (SF), for one, is a way of achieving a glassy state inside and within close vicinity of cells. (Cells cannot live in ice without a glassy border between them and the ice, nor can they live with ice inside them.) Another method is equilibrium vitrification (E-VF) with large amounts of exogenous thickeners (vitrification agents or VFAs).
PRODUCT FOCUS: CELL THERAPIES, BIOLOGICS
PROCESS FOCUS: F
WHO SHOULD READ: PROCESS DEVELOPMENT, MANUFACTURING, QA/QC
KEYWORDS: LYOPHILIZATION, CRYOPRESERVATION, BIOSTABILIZATION, VITRIFICATION, DRYING
LEVEL: INTERMEDIATE
Five Basic Methods of Long-Term Biostabilization
Five major ways of cell stabilization lead to low- or high-temperature VF of intracellular milieu (3,4): high-temperature vitrification, slow freezing to moderaly low temperatures, equilibrium freezing, ice-free equilibrium, and intracellular ice-free kinetic freezing.

One method that forms no ice is the high-temperature vitrification of a highly dehydrated sample (desiccation) and its stabilization by air or vacuum drying at temperatures >0 °C. Sometimes this method is called “lyopreservation,” but that is incorrect because “lyo” implies sublimation. (Lyo is from the Greek word luein, which means to loosen; thus, loosening ice during sublimation.) Xeroprotective agents such as trehalose are used to prevent damage associated with high levels of dehydration. In such cases, those agents are used during secondary drying, freeze-drying, and the whole desiccation cycle. The temperature of drying Tdr is always above the sample's glass-transition temperature Tg for both methods. Otherwise, neither sublimation nor evaporation will occur because of a sample's extremely high viscosity. For stable storage, the temperature of storage Tst must be below Tg. So the conditions of stable drying are Tst < Tg < Tdr(f), where Tdr(f) is the final temperature of drying.
Much of the literature about drying biologicals reports Tg above Tdr, which is incorrect, and we have provided examples and explanations of such a “paradox” (3,5). Such studies show instability of samples at long storage (6) and argue against claims that Tg is within 60–70 °C. In fact, Tg barely exceeds 0 °C (or is within the negative range), so samples cannot be stored long-term at ambient temperatures.
Slow freezing to moderately low (–60 °C to –40 °C) temperatures consists of primary drying and sublimation of the bulk of ice at very high vacuum temperatures. This method is called lyophilization and is widely used in food production, microbiology, and in the biopharmaceutical industry. So far, however, it has had very limited applications in the preservation of animal cells and higher plants.
Equilibrium (slow) freezing allows the freeze-out of the bulk of both extracellular and intracellular water to ice. Water escapes from the cell as the extracellular liquid phase becomes more and more concentrated. Cells are then vitrified in the “inter-ice channels” that are surrounded by ice. The Tg value in those channels is low, however, so cells must be stored in liquid nitrogen (LN2) at –196 °C, in nitrogen vapor, or in industrial freezers at –130 °C. They may be stored for limited times at higher temperature than the Tg of water (around –136 °C), for example, in more accessible –80 °C freezers. This is the mainstream conventional cryopreservation, which in most cases requires the use of permeable and impermeable cryoprotective agents (CPAs).
Ice-free equilibrium vitrification (E-VF) of cell suspensions, tissues, and organs at very low temperatures and moderate to high rates of freezing requires the use of high concentrations of vitrificants. That elevates the viscosity of the milieu and prevents ice formation during cooling and devitrification during warming. Some researchers (7) refer to this method as “vitrification proper,” but in its “pure form” it has had very limited success in preserving animal oocytes, embryos, some tissues (and one organ), as well as some plant specimens.
Intracellular ice-free kinetic (K-VF) vitrification of a bulk solution requires very fast (abrupt) plunging into a cooling agent such as liquid nitrogen. The extremely high rate of cooling (104–106 °C/min) and practically instant warming prevents ice formation inside cells. Ice still can form outside the cells, but it has no time to cause any osmotic damage to cells because K-VF occurs in fractions of a second. Because this method does not require potentially toxic high concentrations of CPAs or permeable exogenous vitrificants, it is also referred as CPA-free vitrification.
The last three methods above involve low temperatures and are in the realm of cryopreservation. Biostabilization >0 °C (the first two methods above) often is considered a part of preservation science and traditionally reported in cryospecialized journals.
Biostabilization Technologies
Xeropreservation Pros and Cons: Biostabilization of noncellular items such as proteins, RNA, and DNA preparations often involves lyophilization and ice-free desiccation. The major issue is not sample survival. Rather, the main concerns are logistic questions, scale-up issues, and so on.
Original attempts to desiccate large quantities of biological solutions faced challenges presented by high viscosities and drastic drops in the rate of evaporation of dehydrated samples. Scientists and engineers circumvented those obstacles by introducing lyophilization, which uses the effect of water sublimation from ice crystals. The rate of that process depends on the level of vacuum and when the noncrystallized phase (cake) can be further desiccated by elevation of the cake temperature >0 °C. Since that introduction, the vaccine industry has made use of giant lyophilizers. But the process is still very slow and consequently has low productivity.
Different means of high-temperature (ice-free) desiccation have been reintroduced recently. Such techniques include droplet evaporation and nebulization (dispersion to small droplets), foaming, and 3D-scaffold surface drying. Those methods are regaining momentum as promising substitutions of the lyophase, but it will probably take many years to completely replace freeze-drying technologies.
The first two desiccation methods above (high-temperature vitrification and slow freezing to moderately low temperatures) have been successfully used for some cellular items, especially microbes, fungi, plants, and some primitive animal eukaryotes. But those techniques have not been useful for cells of high animals, especially vertebrates in general and mammalian cells in particular. Despite the reports of “successful” xeropreservation and lyophilization of live vertebrate cells from time to time by many groups since the end of 1940s — with three notable reports from Meryman and the birth of a cow called “Desicca” (6) — the methods to date have not been proven to produce stable and viable cells that could be stored for a long time. The fact is that even for “good” vitrificants such as proteins, achieving a true high Tg coincides with very low water content (in a range of 0.3 g H2O per 1 g dry weight), which apparently is not sustainable by vertebrate cells so far. Whether other reports will change the situation remains to be seen (8,9,10).
A new approach has been expressed in some studies (5,11,12). It should be noted that sometimes reported “successful” (bringing offspring) freeze-drying or drying of spermatozoa can be a confusing statement. Any properly freeze-dried/desiccated spermatozoa are dead, they are never motile. It is the genetic apparatus, including such excellent endogenous vitrificants as proteins (e.g., histones) and at lesser extent DNA — that can be stabilized at high temperatures (>0 °C) for longer periods of time using xeropreservation or lyophilization.
Slow Freezing (SF): Still the Mainstream of Cryopreservation? The discovery of “enigmatic glycerol” (13,14) led to the explosion of methods of cryopreservation and to the types of species cryopreserved. It also facilitated development of the first cryobanks in the 1950s and revolutionized first the cattle industry and then blood transfusion. Although many applications were successful, the method per se remained semiempirical.
That changed, however, with introduction of the two-factor hypothesis and equations for equilibrium slow freezing (minimal intracellular ice formation) (15,16). Using a truly fundamental approach, Mazur and colleagues in the United States and Wilmut in the United Kingdom were able to cryopreserve mouse embryos (17,18). Since then, slow freezing has been the mainstream of modern cryobiology, and although VF is an emerging method that may one day replace SF for many types of cells, it has not done so yet. Currently, SF is imperative for the majority of cell types.
With development of Mazur's equations, the two-factor hypothesis of cryodamage (15,19,20), and the work of other cryobiologists on slow (equilibrium) freezing in the 1960s, it became clear that each cell type would need its own optimal cryopreservation protocol. That would largely depend on cryobiological and physiological parameters as well as on the type of cryoprotective agents (CPAs) used (21). Particularly, equilibrium freezing of embryos would require a very slow pace of cooling (0.3–1 °C/min) so the cryopreservation process would take several hours. In contrast, for small oblate (flat) ellipsoids such as red blood cells (RBCs) with an excellent surface-to-area ratio that would allow them to lose water very quickly, the optimal freezing rate of cooling would be in the range of several thousand °C per minute. So if we consider an intermediate cooling rate such as 10 °C/min, then it would kill oocytes at a very fast rate because of intracellular ice formation (IIF). But that same cooling rate is too slow for RBCs, so they would die because of excessive shrinkage and prolonged degradation (“solute effects”). Yet for lymphocytes (intermediate between oocytes and RBCs), that rate would be optimal. In addition, the optimal CPA concentration (more precisely, the combination of concentration of CPA and rate of cooling) is unique for a particular type of cell.
SF often requires elaborate multistep protocols that require special equipment. It can do exceptionally well, especially if combined with other “tricks” that are specific to a particular species of cells. A good example is the cryopreservation of human pluripotent (embryonic and induced) stem cells (hESCs and iPSCs). The introduction of a multistep cooling protocol, Rho-associated protein kinase (ROCK) inhibitors, in combination with freezing pluripotent stem cells in adherent stage as they are prone to anoikis (cell death after cells are detached from an extracellular matrix) (22) have dramatically increased survival and functionality of human pluripotent cells after SF (23,24,25,26,27,28,29). SF now highly supersedes various vitrification techniques proposed from time to time (30,31,32).
However, the strengths of SF can be its weaknesses as well. It needs elaborate protocols that have to be developed separately for each new species of cells. It is difficult to implement for CP of large chunks of tissue, and, especially, for CP of a whole organ. But methods and equipment in development (33).
Cryopreservation of Tissues and Organs
One challenge in cryopreserving biological samples is the suppression of damaging ice formation. With over 70,000 total knee replacements required in the United Kingdom every year, availability of living cartilage for knee repair would be a major benefit for patients and the health services industry. So ice formation in human articular cartilage is of particular importance. Although chondrocytes (the single cell type present in cartilage) can be easily cryopreserved in isolation, conventional cryopreservation causes ice formation in the cartilage matrix and chondron. That can cause irreparable damage to the tissue. Vitrification using conventional techniques requires very high cooling and warming rates, which are impossible to achieve accurately in dense cartilage matrix in a clinical laboratory. The technique of Automated Liquidus Tracking offers an opportunity to use practical cooling and warming rates balanced with gradual increases and decreases in cryoprotectant (CPA) concentration (The photo above shows research associate Dr. Alasdair Kay using the experimental system for tissue vitrification). Ice formation can therefore be suppressed while simultaneously minimizing cryoprotectant toxicity across the full temperature range, from bench to liquid nitrogen storage. Successful cryopreservation of articular cartilage represents an excellent stepping stone toward achieving cryopreservation of more complex tissues and organs.

Equilibrium Vitrification and Biostabilization of Large Organs and Tissues: Cryogenic vitrification can be achieved using two basic and reciprocal methods. One is by raising the concentration — and as a result the viscosity — of the intra- and extracellular milieu at relatively moderate and even slow rates of cooling with very high concentrations of VFAs. This method is defined as equilibrium vitrification (the fourth method described above).
Researchers have reported vitrification of a whole organ (kidney) (34) and E-VF of mouse embryo (35). Since those studies, the fate of VF of those two types of cells has split dramatically. E-VF of a whole organ reportedly has had very little progress to date (36).
The second method is by increasing the rate of cooling with few or no exogenous VFAs present. This technique does not cause deleterious intracellular ice formation because of the lack of time available for growing ice crystal nuclei. It is known as kinetic vitrification (the fifth method described above).
Modern methods for vitrification of reproductive, stem, and other germplasm cells are in the realm of kinetic vitrification. The fate of the second direction of vitrification has been different: It has not been “stuck in the mud.” The use of vitrification for cryopreservation of reproductive cells and tissues has boomed during the past 20 years since that seminal paper was published. However, modern methods of vitrification for oocytes, embryos, sperm, and ovarian and testicular tissue are the varieties of kinetic vitrification. Researchers have developed elaborative multistep protocols — involving the addition of VFAs before vitrification and elution of them after warming — to decrease the toxic and osmotic effects of vitrification (37). As Figure 1 shows, numerous carriers also have been developed as well (38).

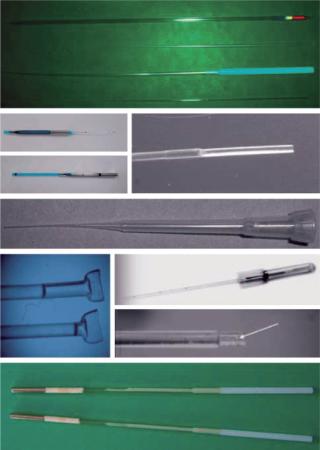
Although a debate remains over whether slow freezing or kinetic vitrification is better for a particular cell type or species, the latter is gaining more approval, particularly for the vitrification of oocytes, thanks to ART clinic scientists and practitioners such as Kuwayama, Vajta, Sheldon, Liebermann, Tucker, and others (37). Although it is faster and simpler than SF (though much farther from automation and not foolproof), kinetic vitrification has been struggling basically with the same problem as SF: For each type of cell, both the carrier and vitrification media need their own protocol. Very often vitrification media that work for open carriers are too dilute for closed carriers. And using open carriers raises the concern of contamination. So kinetic vitrification often has to be checked and adjusted when a new type of cell or species are considered.
Another difficulty with K-VF is that it needs very high cooling and warming speeds. Those can become very difficult tasks for large sample sizes and high production speeds. My company's experience with vitrification has led us to conclude that such difficulties might be overcome soon, and kinetic vitrification might become a mainstream of a biostabilization approach in many areas of reproductive and regenerative medicine and other applications.
Emerging Approaches to Cryogenic Biostabilization
Kinetic Vitrification: Scale-Up and Bioprocessing Issues: Evidently, kinetic vitrification is dominating the present art of vitrification. All efforts are moving in the direction of increasing speeds and decreasing concentrations. However, “traditional” small-size vitrification systems (Figure 1) used in reproductive biology and CP of small amount of cells and/or sample size do not fit other fields that require large samples sizes and fast automatic production.
Another challenge is the cryogenic Leidenfrost effect (LFE), which is a formation of a vapor thermoinsulative coat around a sample placed in a liquid cryogenic coolant such as liquid nitrogen. Novel systems for kinetic vitrification have been developed, including using “cryo slush” (39), cryogenic oscillating heat pipes (40,41), thin film evaporation on a microstructured surface (42), nanodroplet ejection (43,44), quartz capillaries (46), and the KrioBlast system (47,48,49). Our analysis (49) indicates that only two — nanodroplets and the KrioBlast system — address both the processing aspects (scaling up and automation) and the elimination of cryogenic LFE.
Ejecting nanodroplets into LN2 using a “cell printer” technique does not produce sufficient cooling rates. Demirci and colleagues (43,45) hypothesized that very small droplets (a fraction of a microliter, or even tens of nanoliters) would not levitate or hover on the surface of LN2 but would immediately sink and vitrify, thereby eliminating cryogenic LFE. We have obtained results that do not support this theory. Our experimental curve “droplet volume–time of Leidenfrost levitation” plateaued earlier than predicted by Demirci (shown as Figure 3E in Reference (44). So even the smallest droplets will hover for sufficient amounts of time. As such, the rate of such small droplet cooling would not exceed tens of thousands °C per minute (50). In addition, the droplet-making device (analogous to the cell printer of a cell sorter ejector) is impractical, especially for the harvesting of micron-sized droplets and the relatively high concentrations of the vitrificants that are still needed, (1.5 M propandiol and 0.25 M sucrose) (48).
From our experience, bull sperm exhibits only 10–20% of normal motility in such a solution. We also thoroughly examined the sources of discrepancy and developed a mathematical model of the droplet cooling system dynamics (50). That model shows that nanodroplets still have a relative long time (1-2 seconds) hovering as a result of LFE and the floating force created by the vapor coat, which would keep the droplets over the top of the surface of LN2 and, thus, prevent higher cooling rates before submerging. Even 1 second of hovering is not fast enough to vitrify very dilute solutions. Our theoretical model is in good concordance with our experimental data (50), but the cooling rates are some three to four times lower than claimed by the Demirci group (43).
A new method has been developed for fast kinetic vitrification of cells in large volumes. Of the methods listed above — except perhaps the “slush” vitrification, which even then has limitations with cooling speeds (33) — each requires a sample be placed into coolant. Katkov and Bolyukh have developed a technique that involves blasting a stream of pressurized liquid nitrogen at the sample. This is analogous perhaps to a firefighter who uses a high-pressure hose to cool hot items. In the same way, industrial devices can be chilled using a high-speed flash of pressurized coolant. To the best of our knowledge, this concept has not been applied in cryobiology to vitrify and preserve cells. The major propositional aspects of this approach along with some potential practical designs are set out in Reference (17).
In 2010, Katkov and Bolyukh built a basic manually operated apparatus called KrioBlast-1 (47). It vitrified up to 4,000 µL with a 15–16% glycerol solution. That corresponds to 100,000– 600,000 °C/min. (The process of vitrifying a 4-mL solution with a clear view of a sample inside the chamber is shown in a video on the website www.celltronxibio.com.)
A second-generation machine was then produced with an automated cooling option (49). The cooling rate was extrapolated from critical cooling rate graphs similar to that shown in previous studies (51). Our studies showed that using KrioBlast-2 equipment with modifications, a 12% solution of glycerol (and in some occasions as low as 7% of glycerol solution) might be stably vitrified. That would correspond to cooling rates of above a million °C/min, accordingly to extrapolation of the Warkentin's plot. Our thermodynamic analysis (50) of heat propagation indicated four- to sixfold lower numbers given in Reference (51), which could possibly indicate an inapplicability of the current theories of glass transition to very dilute aqueous solutions. Preliminary results with cells showed very high (90–95%) recovery of vitrified human embryonic stem cells and spermatozoa in cases when warming was sufficiently fast. However, if warming (performed manually) was slow, survival was very low (5–10% for hESCs, slightly higher for spermatozoa). Current work is proceeding on the development of a third generation of the device that would permit both hyper-fast cooling and hyper-fast warming and could lead to an automated scaled-up product.
Future Advancements
The methods proposed by the Demirci group and Celltronix are in the stages of active development. So only the future will show which, if either, could be more applicable to bioprocessing needs or biostabilization in larger scale and usable with more types of cells. However, it may well be that other proposals and methods emerge. As I forecasted some three years ago, “A Race for the Pace” is occurring in the search for a “universal cryopreservation” platform — or at least one suitable for single, suspendial cells and thin tissue samples.
Author Details
Igor I. Katkov is the chief scientific officer at Celltronix, San Diego, CA, 1-858-577-0211;
REFERENCES