Disposable bioprocessing has come of age. Economic and regulatory conditions are driving the widespread adoption of disposable equipment at all stages of bioprocessing. This review considers the entire bioprocess chain and assesses the status of disposables. In particular, we focus on the current availability and need for additional sensors that will enable the disposable process to be integrated — in compliance — with the latest Process Analytical Technologies.
Traditional bioprocessing is highly compartmentalized into upstream and downstream operations. Within these broad divisions, one finds early stage, pilot and manufacturing units as further subdivisions. In an ideal world, there should be a seamless handoff from discovery through to manufacturing. In reality, many companies operate with departments that end up being vertically integrated, but not horizontally. The rising costs of drugs and economic pressures are creating a “perfect storm.” Coupled with the sequencing of the human genome, there is now a tremendous opportunity to pursue novel biologics for a variety of disease states. However, the ballooning costs of getting a drug to market ensures that only a few companies have the resources to make it. In this environment, providing lower-cost bioprocess solutions without compromising on GMP capability are highly desirable. An additional benefit of having a low-cost disposable bioprocessing capability would be the ability for governments to respond to potential natural or bioterror infectious diseases.
The market for disposables is growing rapidly. The obvious advantages of cutting out the expensive installation and validation costs associated with sterilizing-in-place and cleaning-in-place are clear. Therefore, many companies have solutions that provide disposables that can be used to replace virtually any unit operation associated with biotechnology manufacturing. Companies using disposables do not need to invest significant upfront capital in equipment purchase, and this factor essentially becomes a consumables cost. However, a major flaw with these disposables is that they have very little process monitoring capability, especially at the culturing stage. Given the profound effect that the cellular environment has on product formation and quality, it is vital to monitor process parameters. The FDA has recognized this and has actively promoted the use of Process Analytical Technology (PAT) (1,2,3).
Upstream Bioprocess Development
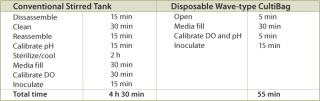
A typical set of operations in any biologic development process is shown in Table 1. As can be seen, the current state-of-the-art only employs instrumented reactors at the lab scale and beyond. This leads to a major technology gap; by the time the culture reaches the stage when it needs to be optimized, it has already been selected out of several hundred possible candidates that were grown in non-instrumented vessels and, thereby, any opportunity to link process conditions to productivity is lost. Fortunately, the recent maturation of non-invasive sensor technology promises to provide a solution to the problem (4,5,6,7). Sensor technology, by its very nature, is independent of scale and can therefore find its way into any disposable process equipment. Figure 1 illustrates how such sensors are typically constructed and used. These sensors have a number of advantages, as listed. In addition, the concept of sensing more than one parameter in a bioreactor is protected by US patents (8,9). These have been licensed to BioProcessors (Woburn, MA, USA) for volumes less than 1 mL and, exclusively, to Sartorius Stedim Biotech (SSB, Aubagne, France) for all other applications. Fluorometrix (Waltham, MA, USA) is focusing on the 50 mL minibioreactor product and on sensors for monitoring shake and spinner flasks. As an exercise, we compared the time and labour taken to conduct a 3 L scale cell culture in a traditional versus a disposable system. Table 2 demonstrates the time savings gained in a single development experiment by using a lab-scale CultiBag wave-type disposable reactor versus a traditional stirred tank, clearly demonstrating the advantage of using a disposable system.
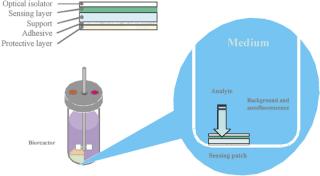
Table 1: Platforms used for upstream bioprocess development.
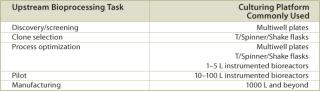
Table 2: A comparison of the steps and time necessary to set up and operate conventional and disposable bioreactors.
Purification Optimization
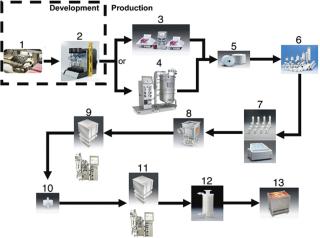
Shown in Figure 2 is a hypothetical process, consisting largely of disposable components available through Sartorius-Stedim, which creates the ability to be (a) flexible within a production process and (b) optimize equipment utilization within an existing process. Existing cation or anion chromatography steps are mainly used for the removal of contaminants, such as host cell proteins, DNA or endotoxins from the product stream. These columns can be used once or in multiple cycles. The set up of gel chromatography columns, including their packing and cleaning, etc., takes time (sometimes up to 8 hours). Additionally, the chromatographic step itself can be extremely lengthy, often taking 32 hours or more, which not only contributes to the overall processing time, but also to the process irreproducibility. Moreover, the capacity utilization of traditional gel bead chromatography columns is not efficient — as the molecules are too large to easily diffuse into the bead structure (see Figure 3). Membrane adsorber technology has shown to be far more effective than the use of gel beads. Figure 3 shows, conceptually, the two different mechanisms of the different technologies. As illustrated, it will be much more difficult for a DNA contaminant to diffuse into the tortuous interior of the gel bead as compared with the membrane adsorber because of the much larger pore structure of the latter.

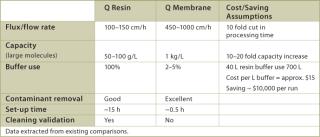
The large pore size of the membrane adsorber not only allows a far higher efficacy of capture, but also decreases the processing time owing to high flow rates. In Table 3, data from existing applications show that process times can be reduced 10–15 fold, and that flush volumes and hold-up volumes are also reduced by a similar factor. Additionally, membrane chromatography is available in disposable or reusable formats; that is, no column set up is required, which can be difficult and time consuming. The data given in Table 3 demonstrate that savings in buffer consumption alone would be substantial, not even including the savings stemming from yield and capacity improvements as a result of reduced process times. Other advantages associated with the use of membrane chromatography include the reduction of cleaning solutions, downtime and labour. Purification process optimization is a complex problem involving many factors related to cost savings and revenue increase. We believe that the use of disposable membrane chromatography technology will greatly increase process economics and that studies of this technology should be considered a high priority project.
Table 3: Performance comparison between Q-resin chromato graphy column and Q-Membrane.
Final Fill: A variety of tanks are commonly used in the final fill process. All tanks require substantial cleaning, set up, WFI, vent filters and footprint. This results in appreciable costs related to downtime, labour, required solutions and capital expenses. The entire system of tank, vent filter and liquid filters could be set up as a disposable system. A disposable bag could be used that has all the required filters connected to it and is gamma radiation sterilized. The end-user simply cuts the packaging open, folds the bag into a holder and aseptically connects the filters to the filtrate side. In other processes that employed a more traditional final fill approach, cleaning, set up and sterilization took up to 6 hours, so that very often at this point the process extended into the next shift or even the next day. Disposable holding bag technology would reduce this timeframe to just unpacking and set up as described above. The advantages listed in Table 4 are the result of a final fill system set up as described.
Table 4: Advantages of a disposable final fill system.
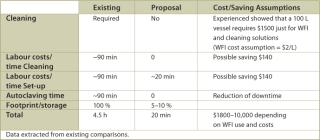
Mixing (Media and Buffers): In some instances, media or buffer mixing vessels require up to 12 hours of CIP to eliminate any contaminant or cleaning agent residue. Similar to the previously described holding bags, the mixing step can be performed with disposable mixing bags, which require minimal set up time and avoid the need for cleaning. As with disposable holding bags, these mixing bags would be presterilized with gamma irradiation. Calculations based on processing comparisons indicate that cost savings derived from using disposable mixing bags for media and buffer mixing are similar to those described for final fill processing.
Other Technologies: Cell harvests with either depth or cross-flow filtration, as well as filtrative sterilization steps, are already done in the industry using disposable, presterilized formats. The cleaning of such systems is again unnecessary and system set up is reduced to a minimum. Innovator companies face long and costly timelines to develop their new pharmaceutical products and eventually obtain regulatory approval from the appropriate regulatory agencies. Although figures quoted depend on the reporting source, it is not uncommon to hear that development approval cycles for a new molecular entity can take between 10 and 15 years, with a total development cost in the $800 million range.
Clinical trials typically include three different stages. Phase I is aimed at an initial safety evaluation, and includes a small (<100) number of healthy volunteers. Phase II continues the safety evaluation and is designed to obtain a preliminary efficacy indication in hundreds of patients. Pivotal efficacy studies are done in Phase III, which includes thousands of patients. In some cases, a post-approval/post-licensure Phase IV clinical study is done at FDA’s request to answer any residual questions. It is FDA’s goal, through this approval process, to ensure, with the best possible methodology, that safe and effective products reach the marketplace. It is the agency’s expectation that a consistent quality product, from lot-to-lot, is obtained from an appropriately controlled manufacturing process. The primary aim of pharmaceutical product development is to design a quality product and a manufacturing process for such items that consistently delivers the intended performance of the product. Product quality is obtained by building it into the design of the product and its manufacturing process (quality by design, or QBD). Thus, manufacturers need to establish what the critical quality attributes (CQAs) are for a certain product to ensure its safety, efficacy, stability and processability. The establishment of CQAs requires a thorough understanding of the product.
Although classical process validation approaches are certainly an effective means of establishing process consistency vis-à-vis potentially variable process inputs, a paradigm shift is occurring with the introduction of the PAT concept to pharmaceutical and biotechnology manufacturing. PAT is a system for designing, analysing and controlling manufacturing through timely measurements (during processing and, if possible, online) of critical quality and performance attributes of raw and in-process materials and processes with the goal of ensuring final product quality. PAT brings in a level of process understanding that goes beyond simple process monitoring and control … or “recipe control” if you will. PAT brings in product understanding, product CQA monitoring, process monitoring, process understanding and process control towards an integrated approach for product quality control. The implementation of PAT will ensure that product quality is monitored and controlled during the manufacturing process, with process decisions being made based on product attributes. Process validation and the implementation of PAT approaches are very much process-and product-specific. The novel sensors being deployed will permit the establishment of PAT strategies heretofore unavailable for disposable bioreactor systems.
REFERENCES