Evaluating single-use systems for extractables and leachables is new territory for many end-users. This paper presents a risk-based approach for evaluating extractables and leachables from single-use systems. Approaches are described that have been accepted by various regulatory agencies utilizing risk assessments and sound scientific principles.
Biopharmaceutical processing materials must be evaluated to determine whether they impact the final drug product with regard to safety and efficacy. For single-use and reusable systems, this usually leads to evaluations that demonstrate compatibility with the process formulations. Many of these evaluations can be performed by the vendor and do not need to be addressed by an end-user. Examples of these are as follows:
-
Mechanical properties such as tensile strength (bags) or maximum pressure ratings (filters, tubing)
-
Gas transmission properties (bags)
-
Minimum water bubble point (filters)
-
Chemical resistance.
A comprehensive list of tests that may be performed for single-use systems has been published by the Bio-Process Systems Alliance (BPSA) (3).
Regulatory Requirements for Single-Use Systems: Extractables and LeachablesSingle-use systems, also referred to as processing materials, must be evaluated for their ability to contribute leachables to the final product, but most global regulations do not specifically refer to this requirement. Both EU and US regulations clearly require that production equipment should not “present any hazards to the product” and should not be “reactive, additive or absorptive” (4,5). The EMEA and the FDA have published guidances that address the packaging of final drug products and the potential for leachables to be extracted from packaging materials (6,7). While some upstream processing materials might have less potential impact on the final drug product, the final packaging guidances provide risk assessment strategies that can be applied to processing materials. Extractables and leachables are defined as follows:
Extractables: Chemical compounds that migrate from any product contact material when exposed to an appropriate solvent under exaggerated conditions of time and temperature.
Leachables: Chemical compounds, typically a subset of extractables, that migrate into the drug formulation from any product contact material, — including elastomeric, plastic, glass, stainless steel or coating components — as a result of direct contact with the drug formulation under normal process conditions or accelerated storage conditions and are found in the final drug product.
Extractables and leachables are most often associated with polymeric and elastomeric materials because of the use of additives to increase stability and aid in the formation of the material components. For example, polyolefins such as polyethylene and polypropylene will usually have one to three antioxidants and/or stabilizers and one or more processing aids such as lubricants or antislip additives.
Impact of Processing Material Leachables on Product Safety and EfficacyScience- and risk-based assessments of leachables present in the final product must determine whether leachables are present at levels that cause safety concerns. The results of leachables evaluations should be reviewed by qualified toxicologists. The goal is to minimize patient safety concerns regarding exposure to harmful leachable compounds. The route of administration (parenteral, topical, inhalation or oral) and dose of the drug product (acute versus chronic) are key factors in the safety assessment. The nature of the process and use of the processing materials are also important factors.
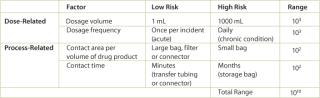
Several risk factors that influence the safety assessment are presented in Table 1. The concentration of a leachable in the final drug product is not as important as the dosage that is accumulated by the patient. The acceptable concentration of a leachable is higher for a 1 mL injection than for a 1000 mL transfusion. In other words, the concentration of the leachable in the 1 mL injection could be 1000 times higher (or 103) and achieve the same dosage of the leachable. For each of the listed factors (dose and process related), low and high risk examples and an estimated range are given. By adding the ranges for each factor, the acceptable concentration of a leachable could vary by as much as 1010 depending on the process and use of the drug product. Therefore, when addressing leachables from processing materials, the concentration measured during a leachables test must be translated to a final dosage level in the drug product.
Table 1: Safety concerns for leachables in final products.
The importance of evaluating extractables and leachables to reduce patient safety risks should not be underestimated. A study published in the PDA Journal of Pharmaceutical Science and Technology (8) explains how replacing human serum albumin (HSA) with polysorbate 80 in a parenteral drug led to a new leachable that resulted in a high incidence of antibody positive pure cell aplasia (PRCA). Although this example is from a final container/closure system, it is an effective demonstration of the potential for leachables to be a serious safety issue. Certain leachable compounds can also affect product efficacy and stability. The end-user must assess the chemical nature and concentration of leachables and their impact on product efficacy. End-users should assemble a team of development and manufacturing scientists, validation specialists, quality assurance and regulatory experts to plan and review the programme. While the resources for extractables and leachables testing can be considerable, the possibility that a comprehensive extractables and leachables programme can prevent safety/efficacy issues and regulatory delays can lead to overall cost savings.
A Risk-Based Approach to Extractables/Leachables EvaluationThe application of risk assessments to pharmaceutical and biopharmaceutical regulatory requirements is recommended by the FDA and other global regulatory agencies. The FDA’s Final Report on Pharmaceutical cGMPs for the 21st Century — A Risk-Based Approach (9) provides clear guidance on utilizing scientifically sound, risk-based approaches to pharmaceutical cGMP requirements. A risk-based programme was suggested by the BPSA to address extractables and leachables from single use systems (10,11). It is highly recommended that this programme be initiated early in the development process when changes are more easily addressed. In the BPSA approach, the extractables/leachables programme begins with and depends on a thorough understanding of the process. Effective science- and risk-based assessments are best done from a foundation of comprehensive process knowledge, which is key to quality by design principles (12). A comprehensive process understanding includes, but is not limited to, research and development studies, process descriptions, batch records, SOPs, technical reports, batch testing, data trending and operating parameters.
Once the process details are understood, a list of product contact materials should include any material that has the potential to migrate into the final product and begins upstream in the process. The list concludes with materials used directly before the final fill of containers. Examples of product contact materials include tubing, bags, filters, connectors, O-rings, syringes, chromatography resins and bulk storage vessels. A risk assessment is then applied to the list of product contact materials to determine which have the greatest potential for leachables. The criteria applied to the risk assessment are specific to the end-user. Figure 1 presents the flow of the risk assessment process. For each product contact material, the following risk factors are evaluated:
-
Material compatibility
-
Location in process
-
Nature of product
-
Surface area
-
Contact temperature and time
-
Pretreatment steps.
Once the relevant risk has been determined for each product contact material, then available extractables data for each product contact material is obtained. If none is available, then the end-user must either generate their own extractables data or proceed directly to leachables testing as shown in Figure 1. If leachables testing is done and no leachables are detected, then the results of the leachable study are submitted in support of the product. If leachables are detected, then they must be identified and quantitated, followed by a toxicological safety assessment.
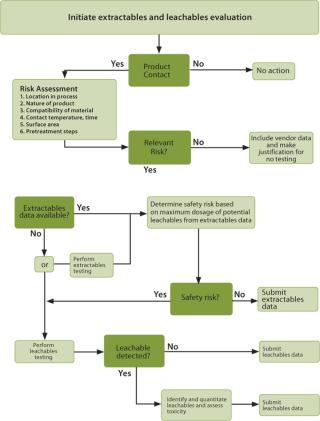
If high quality extractables data is available from the vendor, then it may be possible for the end-user to determine the potential for those extractables to be present as leachables in their final product. High quality extractables data is generated from a test utilizing extreme conditions with respect to time and temperature, a variety of solvents and analysis by multiple methods that can separate, detect and identify individual extractables. The result is a comprehensive list of compounds that have the potential to become leachables. The extent and quality of extractables information from vendors varies and should be carefully reviewed to determine if the conditions used are applicable to the specific situation. If the data is high quality, the concentration of potential leachables is acceptable and it is a low risk situation, then the extractables data may suffice. The use of vendor-supplied extractables data to predict leachables uses the least resources as no new testing needs to be conducted.
Application-Specific Leachables EvaluationsWhen extractables data is not available, not of high quality or not relevant to the specific process situation, then the end-user must perform an application-specific leachables evaluation. There are four choices with which to address leachables, as shown in Figure 2.
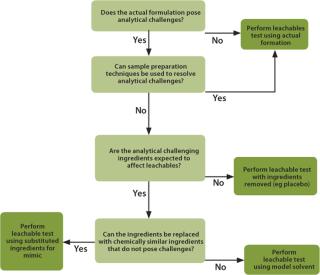
Testing with the Actual Drug Formulation: Testing for leachables with the actual drug formulation is the most direct method to test for leachables and is most often used for final container/closures evaluations. The actual formulation includes processing solutions and intermediates such as media, buffers and product-related solutions used in biopharmaceutical applications. Testing with actual formulations directly satisfies the leachables requirement. The results are used by the end-user to evaluate the impact on the final product. When testing with an actual formulation, it is possible that a leachable will not be detected because of interference from formulation ingredients. This is commonly addressed by using multiple analytical methods that rely on different mechanisms to separate the leachables from the drug formulation. There are situations when standard methodologies are challenged by some or all of the formulation ingredients — most often the active pharmaceutical ingredient (API). In this case, the interference can often be removed by standard sample preparation methods such as solid phase extraction or liquid–liquid extraction. Protein-based ingredients, often found in biopharmaceutical formulations, may also be removed by precipitation.
Removal or Replacement of Ingredients: In applications when the analytical interference cannot be removed, alternatives to testing with the actual formulation must be considered, such as testing with slightly modified actual formulations. If the active pharmaceutical ingredient (API) is causing interference, then removing the API should be considered. Testing placebos, in most cases, is acceptable and provides an almost direct answer to the leachables question. Testing placebos is also applicable when the API is cytotoxic or extremely expensive. A scientific justification must be made to support this approach, which includes evaluating whether the removed component could impact the leachables result. When an ingredient other than the API is causing the analytical interference, it may be possible to remove it from the formulation without impacting the scientific validity of the extraction. Alternatively, the interfering ingredient can be replaced by a compound that does not cause analytical challenges but has similar extraction properties to the original ingredient. For example, replacing benzyl alcohol with n-propanol would be an acceptable modification.
Mimic Solutions: If the major ingredients of the formulation or intermediate solution are causing the analytical challenges, then a complete replacement with a mimic solution can be considered. The mimic is chosen to have similar extraction properties to the actual formulation. The testing is performed using “worst-case” process conditions.
Modelling: In a situation when the actual formulation cannot be analysed or substituted using a placebo or mimic, then a process modelling approach should be considered. Modelling would consider the process parameters and use a suitable solvent or combination of solvents, such as ethanol and/or water to do the modelling evaluation.
Analytical TechniquesThere are numerous analytical techniques used to detect, identify and quantitate extractables and leachables. It is recommended to use a combined analytical approach to perform a comprehensive evaluation of extractables and leachables. The combined approach would utilize at least two specific analytical techniques and may include non-specific techniques. “The necessity of using multiple techniques for the detection, identification and quantitation of the leachables … indicates that no single method is sufficient and suggests that multiple, orthogonal techniques be routinely employed” (8).
Specific Analytical Techniques-
High Performance Liquid Chromatography (HPLC) is considered to be a universal separation tool for organic compounds and works on a broad range of compound molecular weights. HPLC can accommodate multiple detection techniques such as Ultraviolet Diode Array and Mass Spectrometry (MS).
-
Gas Chromatography (GC) is used for semivolatile and volatile analytes and also accommodates multiple detection techniques such as MS, Flame Ionization detectors (FID) and others. GC-MS provides spectral information facilitating the identification of compounds.
-
GC with Headspace Sampling analyses volatile compounds that are vaporized. This technique can be useful when residual solvents are likely to be present in the processing material and also can be used to meet the requirements of USP <467> Residual Solvents (13).
-
Inductively Coupled Plasma (ICP) Spectroscopy is used to screen for a wide range of metal elements.
-
Total Organic Carbon (TOC) analysis may provide estimates of total extractables and leachables for aqueous formulations.
-
Fourier Transform Infrared. Spectroscopy (FTIR) may also provide a total extractables estimation and detect potential extractables that are not otherwise detected by other methods (such as oligomers).
-
Non-Volatile Residue Analysis (NVRA) can provide an estimate of the mass of semi- and non-volatile extractables.
Specific analytical techniques are typically used for the identification and further quantitation of extractables and leachables. Quantitation is generally performed using the analytical technique wherein the extractable or leachable was detected and using a validated method specific to the sample matrix of the actual formulation. Challenges for identification and quantitation may arise because of the sample matrix, the availability (or lack thereof) of standard analytes for identity confirmation, and the potential for the leachables to be degradants of the polymer additive, making systematic identification steps difficult.
ConclusionEvaluating processing materials for extractables and leachables requires co-operation between vendors and end-users and a commitment to utilize the best science possible within a risk-based framework. End-users may find themselves customizing a leachables test to meet their specific applications because other options do not provide a sufficient solution to the requirement. With vendor co-operation and analytical expertise, an application-specific leachables evaluation may prove to be straightforward and successful.