
SARTORIUS STEDIM BIOTECH (WWW.SARTORIUS-STEDIM.COM)
Determination of extractables and leachables for disposable manufacturing systems must be addressed as part of process validation when single-use technology is used. The idea that compounds leach into pharmaceutical formulations or process fluids (e.g., buffer solutions and bulk storage) from processing and storage materials is not new or even unique to plastics. All materials have extractables and potential leachables. When properly evaluated, these are easily addressed and rarely lead to disqualification of a disposable component. Ideally, processing methods and equipment are chosen early in the development lifecycle of a pharmaceutical product. The choice should be made by a dedicated team of scientists, quality assurance and/or regulatory affairs (QA/RA) representatives, and validation specialists working in partnership with component and system suppliers.
It is important first to understand the distinction between extractables and leachables. In recent years, industry and regulatory agencies have reached a consensus in concept on the following general definitions:
Extractables: Chemical compounds that migrate from any product-contact material (including elastomeric, plastic, glass, stainless steel, or coating components) when exposed to an appropriate solvent under exaggerated conditions of time and temperature.
Leachables: Chemical compounds, typically a subset of extractables, that migrate into a drug formulation from any product contact material (including elastomeric, plastic, glass, stainless steel, or coating components) as a result of direct contact under normal process conditions or accelerated storage conditions. These are likely to be found in the final drug product.
The distinction is important to understand. Extractables are determined by exposing components or systems to conditions that are more severe than normally found in a biopharmaceutical process, typically using a variety of solvents at high temperatures. The goal of an extractable study is to identify as many compounds as possible that have the potential to become leachables. A positive outcome is one where the list of extractables from a material is sizable. Although it is not expected that many of those extractables will actually leach into the drug product at detectable levels, a materials extractables profile provides critical information in pursuit of a comprehensive leachables test.
However, it is important to note that not all leachables may be found during the extractables survey (Figure 1). For instance, drug formulation components or buffers may interact with a polymer or its additives to form a new “leachable” contaminant that was not previously identified during extractables analysis. In addition, leachables that were not identified as extractables also will be found if the drug product formulation and processing conditions are unique and more severe than the conditions at which extractable tests were performed — or when the analytical methodologies used in the two types of studies are different.
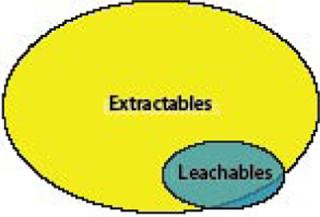
There are as yet no specific standards or guidances that reference extractables and leachables from single-use (disposable) bioprocessing materials. Many references that do apply were written to address all processing materials and equipment without regard to the materials of construction. But it is clear that they are sufficiently broad to include leachables.
North American Requirements (United States and Canada): The foundation for the requirement to assess extractables and leachables in the United States is introduced in Title 21 of the Code of Federal Regulations (CFR) Part 211.65, which states that “Equipment shall be constructed so that surfaces that contact components, in-process materials, or drug products shall not be reactive, additive, or absorptive so as to alter the safety, identity, strength, quality, or purity of the drug product beyond the official or other established requirements” (1). This regulation applies to all materials including metals, glass, and plastics.
Extractables and leachables generally would be considered “additive,” although it is also possible for leachables to interact with a product to yield new contaminants.
The US Food and Drug Administration (FDA) regulatory guidance for final container–closure systems, though not written for process contact materials, gives direction about the type of final product testing that may be provided regarding extractables and leachables from single-use process components and systems (2). The May 1999 guidance document from the FDA’s Center for Drug Evaluation and Research (CDER) indicates the types of drug products and component dosage form interactions that the FDA considers to be the highest risks for extractables (Table 1). Drugs that will be administered as injectables or inhalants will have higher levels of regulatory concern than oral or topical drugs. Similarly, liquid dosage forms will have higher regulatory concern than tablets because extractables migrate into liquids more easily than into solids.
Table 1: Examples of packaging concerns for common dasses of drug products

In addition, pharmaceutical-grade materials are expected to meet or exceed industry and regulatory standards and requirements such as those listed in the US Phar < ill/ > (USP) chapters <87> and <88> (3, 4). The USP procedures test the biological reactivity of mammalian cell cultures following contact with polymeric materials. Those chapters are helpful for testing the suitability of plastics for use in fabricating a system to process parenteral drug formulations. However, they are not considered sufficient regulatory documentation for extractables and leachables because many toxicological indicators are not evaluated, including subacute and chronic toxicity along with evaluation of carcinogenic, reproductive, developmental, neurological, and immunolog
ical effects.
Polymer formulations approved for contact with foods have a list of allowed compositions and additives specifically intended to limit extracted or transferred substances. To the extent that such regulations limit composition, they also allow some prediction of extractables. In the United States, these listings are complex and difficult to use because allowable additives vary by intended use. European Union food-contact requirements are narrower and better defined. However, the predicted extractables profile still should be confirmed. Use of food-contact–listed materials cannot be substituted for determination of extractables.
International Requirements: In the European Union, a related statement to the US 21 CFR 211.65 is found in the rules governing manufacture of medicinal products. The EU good manufacturing practice document states that “Production equipment should not present any hazard to the products. The parts of the production equipment that come into contact with the product must not be reactive, additive or absorptive to such an extent that it will affect the quality of the product and thus present any hazard” (5).
The European Medicines Evaluation Agency (EMEA) published a guideline on plastic immediate packaging materials in December 2005 that also addresses container–closure systems and has been used to provide direction for single-use process contact materials (6). Extractables and leachables data that must be submitted depends on the nature of an active substance (e.g, solid or liquid) and the route of administration (e.g, inhalation, parenteral, ophthalmic, oral, or topical). If a plastic material is described in the European Pharmacopoeia (Ph. Eur.), the level of information necessary for submission is lower. For “nonsolid” active substances, the general requirements include the complete qualitative and quantitative composition of a given plastic material. Data to be included relating to extractables and leachables come from extraction studies (“worst-case leachables”), interaction studies, migration studies (similar to leachable studies), sorption studies (interaction between drug formulation and packaging), and toxicological information/documentation.
A GMP guide from the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) considers the impact of equipment on the final product by stating that “Equipment should be constructed so that surfaces that contact raw materials, intermediates, or APIs do not alter the quality of the intermediates and APIs beyond the official or other established specifications” (7).
Industry Guides: Extractables and leachables have been addressed specifically for filters, which gives an indication what might be expected for all disposables. The Parenteral Drug Association (PDA) published a technical report to guide users with the selection and validation of liquid-sterilizing–grade filters (8). In addressing extractables, it specifies that “It is the user’s responsibility to demonstrate that the product does not contain objectionable levels of extractables from the filter.… The filter user is responsible for obtaining extractable data for the drug product formulation” (8).
The Product Quality Research Institute (PQRI) is a collaborative process that includes members of the US FDA, industry, and academia. In 2006 it undertook a substantial effort to define the appropriate testing procedures and safety evaluations to qualify final inhalation containers for orally inhaled nasal drug products (OINDPs) with regard to extractables and leachables (9). OINDPs are considered high-risk products (Table 1), so the PQRI recommendations are not meant to be applied to container–closures for other products such as parenterals — or for biopharmaceutical processing materials. However, the PQRI tested custom formulations of polypropylene resin, sulfur-cured, and peroxide-cured elastomers. Extraction techniques and analytical methods were investigated that advanced the science and can form a basis for successful extractables and leachables programs.
Initiating an Extractables and Leachables ProgramKnow Your Process: The first step of initiating an extractables and leachables program is to understand your manufacturing process completely from start to finish. A detailed process description is usually derived from research reports, batch records and associated standard operating procedures (SOP), technical reports, and batch analyses, all tools to use in preparation for mapping the process. Discrepancies between those documents should be resolved so that an accurate representation of the process is made. For example, the actual time it takes to fill a bag through a filter may be different from what was originally depicted in an SOP.
Product Contact is defined as contact with any liquid that has the potential to be included in a final product — not only the finished product. This absolutely includes all liquid contact for starting materials and process intermediates. It also may include materials that contact gases or solids, depending on the chemical interactions involved. Materials that may have product contact include but are not limited to
-
Tubing and connectors used to transfer starting buffers into a process stream
-
Syringes and needles used to transfer liquids
-
Sterile and nonsterile filters used to filter the starting buffers and intermediate process formulations
-
Bags and tubing used to store and transfer intermediate process formulations
-
Tangential-flow (cross-flow) filters used for concentration and diafiltration of process intermediates
-
Column chromatography sorbents and chromatography membranes used for concentration and purification of process intermediates
-
O-rings used to seal connectors and sanitary fittings.
-
Final bulk containers (including closure materials of construction) and filters used before final filling.
If there is no product contact as defined above, it is unlikely that further analysis needs to be performed for a given item. For instance, a connector may have components that are not exposed to an actual product stream, and they may not have to be addressed in an extractables/leachables assessment. However, it is important to note that not all materials need to be in direct contact with a drug product to be of concern. Some extractables and leachables may migrate through a direct-contact layer. An example would be a bag with multiple layers that contain differing materials. Extractables and leachables from the outer layers may migrate into a pharmaceutical formulation. Similarly, adhesives and inks from labels may migrate through the layers of a bag.
Single-use systems often combine individual components into a unique, customized package. For example, the bag system illustrated in Photo 1 could have a bag from Vendor A, tubing from Vendor B, connectors from Vendor C, and a filter from Vendor D. The challenge of an extractables and leachables program is to collect and organize all the available extractables and leachables information for those components, identify what additional information or testing is required, and then set and execute a plan to fill in the gaps.
DEFINITIONS
Fourier-Transform Infrared Spectroscopy (FTIR): An analytical technique in which samples are subjected to infrared radiation. Infrared spectroscopy can supply functional-group information and is useful for identification of unknowns. This technique is most valuable for relatively pure compounds.
Gas Chromatography (GC): An analytical separation technique in which samples are vaporized into an inert carrier gas and pa
ssed through a capillary column containing a stationary phase. The column separates individual components of the sample based on intrinsic chemical properties such as molecular weight, polarity, and vapor pressure. GC can detect most compounds as long as they are volatile or semivolatile.
High-Performance Liquid Chromatography (HPLC): An analytical separation technique in which samples are injected into a polar or nonpolar aqueous or organic solvent mobile phase and carried through a column containing a stationary phase. The column separates components of the sample based on chemical properties such as molecular weight, polarity, and electrical charge. Those components are detected by one or more methods such as UV-vis (ultraviolet–visible light), mass spectrometry (MS), refractive index, and fluorescence. HPLC can separate compounds regardless of their volatility. A limitation is that no single detector can detect all molecules.
Inductively Coupled Plasma (ICP) and ICP-MS: Analytical techniques by which a sample is nebulized into a gaseous plasma with its molecules decomposed into atoms. The excited state of each atom emits a particular wavelength of light that can be detected either optically or by MS. Although ICP is amenable to all analytes, it is most commonly used to identify and quantitate metals.
Mass Spectrometry (MS): Detection method that breaks a target compound into ion fragments by applying an electric charge. Those ions are then separated based on their mass and charge. MS can be used as a detector for HPLC (LC-MS) or GC (GC-MS), and an analysis of fragment patterns can help to identify compounds.
Nonvolatile Residue (NVR) Analysis: A method to estimate total extractables in which an extraction media is evaporated to dryness, and the residue is weighed and compared with that of a negative control (solvent that does not contact a test article). NVR measures nonvolatile extractables and many semivolatile extractables. It will not capture volatile and certain semivolatile extractables, depending on the relative vapor pressure difference of the extractables and extraction media. This technique is used with extraction media that do not contain significant nonvolatile compounds.
Product Contact: Contact of a solid with a liquid or semisolid processing medium or dosage form in which such contact could result in migration of leachables that may be included in a final product.
Total Organic Carbon (TOC) Analysis: A method to estimate total extractables in which the organic carbon in extraction media is oxidized to form carbon dioxide, which is then measured using an IR detector. TOC can provide an accurate measurement of organic extractables when used with test media that do not contain significant amounts of carbon.
Once all materials that have product contact have been defined, an extractables and leachables risk assessment can be performed. People evaluating the importance of detailed extractables and leachables data should consider certain risk factors. They include materials compatibility, proximity of components to a final product, composition of the product stream and formulation, surface area of the product-contact components, contact time and temperature, and pretreatment steps.
Compatibility of Materials: Most biopharmaceutical formulations are aqueous-based and therefore compatible with the materials used in most disposable processing components. Still, a check to make sure that the process stream and/or formulation does not violate any of the manufacturer’s recommendations for chemical compatibility, pH, and operating pressure/temperature is warranted before proceeding. A full analysis of data generated by the vendor should be completed up front as a preparatory step.
Proximity of a Component to the Final Product: Product contact immediately before the final fill increases the risk of leachables in a final product. Alternatively, tubing or connectors used to transfer starting buffers probably present a lower risk because of their upstream location. Processing steps such as diafiltration or lyophilization that could remove leachables from a process should also be considered because they may reduce associated risk. However, it cannot be assumed that a step that can potentially remove some leachables will remove all leachables. In such cases, supporting data should be obtained.
Product Composition: In general, a product stream or formulation that has higher levels of organics, particularly high or low pH, or solubilizing agents such as surfactants (detergents), will increase the regulatory and safety concern for potential leachables. Neutral buffers lower concern about potential leachables.
Surface Area: The surface area exposed to a product stream varies widely. It is relatively high for filters, in which the internal surface area is 1,000× the filtration area. Conversely, surface area is relatively small for O-ring seals.
Time and Temperature: Longer contact times allow for more potential leachables to be removed from a material until equilibrium is reached. Higher temperatures lead to more rapid migration of leachables from materials into a process stream or formulation.
Pretreatment Steps: Sterilization by steam autoclave and/or gamma irradiation may cause higher levels of extractables and leachables depending on the polymer formulation involved in a single-use component. On the other hand, rinsing may lower the concern for extractables and leachables (e.g., when filters are flushed before use).
One risk-assessment approach was published by the ad hoc Biopharmaceutical Process Extractables Core Team after summit meetings held in 2002 (10). Priority is established by assigning numerical risks to each category in ranking materials according to their degree of concern. Numerical risks should have supporting data to justify their numerical assignments. If such data are not available, it would be prudent to assign qualitative risks such as “high” or “low” to allow for more general categorization of risk.
Risk assessment is probably the most important part of extractables and leachables evaluation. Each team must evaluate for itself the level of data collection and testing required to address the regulatory and safety risks for its product-contact materials. Some users are comfortable focusing only on the process steps immediately upstream of the final fill. Others will want to perform extractables and/or leachables testing for product contact surfaces throughout the process. It may be that one risk category (e.g., a product formulation with high organic solvent content) is considered of sufficient risk that a more in-depth evaluation of extractables and leachables for it is appropriate without regard to the other risk categories.
Frequently Asked QuestionsExtractables and leachables evaluations are part of a validation program for processes using disposable biopharmaceutical systems and components. All materials have extractables and leachables. There is minimal regulatory guidance that directly addresses extractables and leachables in bioprocessing. Therefore, regulatory and safety risks are minimized by addressing extractables and leachables early in a validation program. The suppliers of single-use systems represent a good place to start to obtain information on extractables. However, it is always the responsibility of a pharmaceutical product sponsor to ensure that leachables and extractables are appropriately addressed.
Who Is Responsible for Conducting an Extractables/Leachables Investigation? Current regulatory responsibility for overall assessment and understanding of a finished product and process components involved in its production remains with the product sponsor. This includes evaluations of extractables and leachables.
Do All Elastomer and Plastic Materials Contain Extractables? Yes,
all elastomeric and plastic-based materials contain extractables specific to the formulated and cured material(s) from which they are constructed.
Are Extractable Contaminants Detected from Stainless Steel Systems? Yes, such contaminants can be in the form of residues left after cleaning or traces of metals such as iron, nickel, and chromium salts from the stainless steel itself.
Why Are Additives Used If They Can Become Extractables or Leachables? Can I Get a Polymer That Does Not Contain These Additives? Most commercial polymers would be nonfunctional without the use of certain additives. These stabilize polymers during processing, lubricate them during extrusion, and prevent oxidation and ultraviolet degradation throughout the shelf life of a polymer. They are also used as antistatic agents, impact modifiers, catalysts, release agents, colorants, brighteners, bactericides, and blowing agents. One exception to this rule is found with fluoropolymers, which are typically processed without additives, stabilizers, or processing aids.
Does a Vendor Need a Drug Master File (DMF) or Biologicals Master File (BMF) to Support My Product? A DMF or BMF for process-contact equipment is not explicitly required by US regulatory authorities. However, it represents a way for vendors to share proprietary information about a component or raw material with the FDA and to ensure that such information remains up to date.
How Often Is DMF or BMF Information Added or Deleted? Once the file has been submitted to the FDA, a vendor is required to update that document annually. Information typical to these files includes a physical description, intended use, chemical composition (including extractables), manufacturing process and locations, and raw material types and grades.
Does the Polymerization Process Used in Manufacturing Affect the Level or Types of Extractable Analytes? Yes, it is now well understood that the choice of polymer or method of polymerization (by heat or chemical means) directly affects the levels and types of compounds found as extractables.
Does the Sterilization Process Used After Manufacturing Affect the Level or Types of Extractable Analytes? Yes, it is now well understood that the levels and types of compounds found as extractable analytes are directly affected by the type and degree of sterilization performed (e.g., gamma irradiation, ethylene oxide gas, or autoclaving).
Can a Supplier Guarantee That No Leachable Will Come Out of a Product That Will Be Deleterious to My Process? Absolutely not. The leachable analyte and concentration that may be of issue to one particular drug formulation may have no impact on another. It is the responsibility of product sponsors to qualify and demonstrate applicability of process components within their manufacturing systems. Leachables are final-product–specific.
What Are the Most Common Ways That Product Sponsors Have Trouble with Extractables and Leachables? It is the responsibility of each product sponsor to fully understand its process, including the potential and possible origins for product adulteration. When a sponsor does not understand (or care to look for) the presence of extractable analytes, it runs the risk of less-than-optimum quality product development, potential safety risks, and probable regulatory delays.
Completing an Evaluation ProgramBefore extractables and leachables evaluation, a process should be reviewed and documented and a comprehensive list made of the processing materials it uses. In addition, key operating information such as formulation composition, processing volumes, and contact times and temperatures should be documented. Other applicable information (e.g., pH, product stability, and viscosity) may also affect the extracting capabilities of a formulation.
Figure 2 shows BPSA’s recommended program for addressing leachables and extractables.
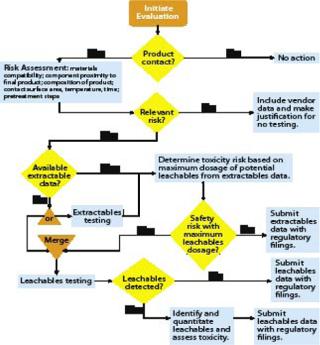
Product Contact: All materials with product contact need to be addressed for extractables and leachables. In review of a materials list for a biopharmaceutical process, it may become apparent that some materials do not have intimate contact with the product. With such materials, extractables and leachables are less likely to be a problem, so they may not need to be addressed.
Risk Assessment: The second part of an extractables–leachables program is assessing the risk of safety or regulatory concerns based on key risk factors: material compatibility, proximity of a component to the final product, composition of that product, component surface area, contact time and temperature, and related pretreatment steps. When a risk assessment is complete, there may be materials that pose no relevant safety or regulatory risk. In such cases, no further action may be required as long as that can be justified. More likely, some further consideration is appropriate.
Vendor-Provided Extractables Data: If further investigation of extractables and leachables is warranted, a user should obtain an extractables profile, either from the vendor or another source. Ideally, this profile should be produced using appropriate methods so that most potential leachables are identified. Comprehensive extractable data for components can reduce the time and resources needed to qualify leachables from the systems where they are used. When comparing supplied extractables data for components constructed of like materials, end users should carefully review the methods used to generate the data. Less rigorous methods may under-represent the actual levels and extent of extractables, and a report describing more extractables may simply come from using more rigorous methods.
The component used for an extractables study should be the same one that will be used in a process, and it should have the same pretreatment steps as is intended for that process. For instance, if a process uses gamma irradiation for sterilization, then the component used for extractable testing should be sterilized by that method.
It is impractical for a vendor to supply extraction data that brackets all specific process formulations or operating conditions. There will be applications in which end users must obtain a unique extractables profile. Such profiles are ideally generated with the following basic components: extractions using at least two solvents, analysis by high-performance liquid chromatography (HPLC) and gas chromatography with a mass spectrometer detector (GC–MS), additional quantitative investigations, and identification and quantitation of extractables.
Extraction with At Least Two Solvents: The solvents used should include water and a low–molecular-weight alcohol such as ethanol or n-propanol. Where appropriate, an organic solvent with the appropriate solubility parameters will help identify additional extractables. Extractions should be performed at relatively extreme time and temperature conditions. However, the solvents or extraction conditions should not be so extreme as to degrade materials to a point at which they are not mechanically functional (e.g., melting or dissolving). Extreme conditions used should be relative to those under which a material is normally used. For example, one normally used at room temperature might be extracted
at an elevated temperature of 50 °C or 70 °C.
Analytical Methods Should Include HPLC and GC-MS: Methods of analysis used for extractables analysis should detect and identify specific, individual, extractable compounds. HPLC with an ultraviolet (HPLC-UV) or mass spectrometer (LC-MS) detector and GC-MS are the most scientifically robust methods for this purpose. When metals are a concern, inductively coupled plasma analysis is widely used, both with and without mass-spectrometric detection (ICP and ICP-MS).
Additional Quantitative Investigations: Methods such as total organic carbon (TOC), ash, and nonvolatile residue (NVR) analysis can be used individually or collectively to estimate amounts of extractable material present and to ensure that targeted methods are not missing a major extractable constituent. For instance, nonpolar compounds without chromophores can be identified using Fourier-transform infrared (FTIR) analysis of nonvolatile residues.
Identification and quantitation of extractables should be as specific as is practical. The identity of certain extractables often can be verified by comparison of results with commercial analytical standards. However, for some extractables such as siloxanes and oligomers of base polymers, precise identification is not feasible because of the large number of closely related isomers and oligomers. In such cases, a general classification can be used. Quantitation of identified extractables is informative, but it does not need to be performed at a high level of precision. This is different from recommendations for evaluating extractables for final containers, closures, and OINDPs, for which analytical and toxicological limits should be set based on a measured level of extractables (11).
If high-quality extractable data are available, a toxicity assessment can be made for leachables based on the extractable profile. This would be considered a worst-case scenario because the concentration and number of extractables are expected to be higher than what will be measured during leachables testing. Being specific to each application, toxicity assessments are the responsibility of end users.
Unlike in such assessments for final container–closures, single-use systems often include a significant dilution of leachables before dosage. For instance, a 10-L bag may be used for 10,000 doses, in which case the mass of leachables as estimated by the mass of extractables would be divided accordingly for a toxicity assessment. It is therefore possible that risk assessment combined with evaluation of an extractable profile will lead to a conclusion that no further action is required. But it is more likely that additional evaluation will be warranted.
If Extractable Data Are Insufficient or Unavailable: Extractable data will not always be available for the wide variety of materials that contact a biopharmaceutical process stream. Sometimes, available data may not appropriately bracket a specific end-user application. And single-use systems are often customized (e.g., a bag from Vendor A is packaged with a filter from Vendor B, tubing from Vendor C, and connectors from Vendor D). It is not reasonable to expect that such systems constructed using every combination of vendors have been tested for extractables.
If data are available for each component, then they can be evaluated individually. If extractable data are unavailable for one or more components, an end user may choose to perform extractable studies on the assembled combination to obtain specific results for that custom-configured system. However, significant resources are needed to detect, identify, and quantitate extractables. To obtain such data may be worthwhile for end users who plan to use such customized assemblies in many applications. However, it may not be realistic without such economies of scale.
In that case, one alternative for end users is to proceed directly to leachables testing. This would not be considered an appropriate route for final container–closures, for which assessments of both extractables and leachables are generally expected by regulatory agencies.
However, for disposable bioprocessing components, leachables testing without prior extractable testing may be sufficient for regulatory submissions. Bypassing the compilation of extractables data is more likely to be acceptable in lower-risk situations (such as the use of processing buffers) and less likely to be acceptable in higher-risk applications (such as formulations containing significant organic content).
LeachablesLeachables are determined based on extractions that closely model actual processing conditions. For example, an extraction is performed using normal processing temperature instead of extreme temperatures. Instead of extracting with a low–molecular-weight alcohol or hydrocarbon, the actual formulation or a suitable model is used. Because leachables testing uses less-extreme conditions than extractables testing does, leachables detected are usually fewer in number and lower in concentration than extractables would be. If leachables are detected, they should be identified, quantitated, and assessed for toxicity. Often, process conditions are mild enough that no leachables are detected by sensitive analytical methodologies, and such results can be submitted to support a “no leachables” conclusion.
Frequently Asked QuestionsExtractables and leachables are part of a validation program for single-use biopharmaceutical systems. All materials have extractables and potentially have leachables. Minimal regulatory guidance directly addresses these for biopharmaceutical manufacturing systems. Therefore, regulatory and safety risks are minimized by addressing extractables and leachables early in a validation program. The degree of testing and analysis is determined by the relative risk that leachables from a single-use processing material poses to a final drug product. In low-risk applications, minimal evaluations may be appropriate. In higher-risk cases, the degree of testing will approach that required for final container–closures.
Suppliers of single-use bioprocess systems are a good initial source for obtaining information on extractables. However, it is always the responsibility of a product sponsor to ensure that leachables are appropriately addressed. Here are answers to some frequently asked questions about leachables and extractables assessment.
Which component materials should be evaluated for extractables? Those component materials should be evaluated that have the potential to come into direct contact with a manufactured drug product during its production process. Also evaluate those materials that have the potential to come into direct contact with patients.
Should I be concerned if many extractables are detected using aggressive extraction conditions? Because controlled extraction studies are designed to generate extractables, the presence of extractables is expected. This does not necessarily reflect the degree and concentration of leachable compound(s) that will be found upon contact with a product stream. Detection of a toxic or otherwise undesirable extractable under aggressive conditions requires testing to ensure that migration to product is below acceptable limits under actual processing conditions.
What are the methods for preparing plastic and elastomer products to look for extractables? Controlled extraction studies need to use vigorous extraction techniques with multiple solvents of varying polarity to fully elucidate the extractable analytes in question. Techniques such as Soxhlet extraction, solvent refluxing, microwave extraction, sonication, and/or acid washing at elevated temperature may be used.
When I conduct a controlled extractable study, do I extract components whole? It is most common and generally acceptable for processing materials to extract components whole. For extractables testing, you may choos
e to maximize the contact surface area by mechanical methods such as cutting or grinding. For leachables testing, it is most applicable to mimic actual process conditions by leaving test components intact.
What are the appropriate solvents for extractable studies? Regardless of the technique(s) used, controlled extraction studies should use extraction media of varying polarities and physical properties. Ideally, you would obtain extractable data from two or three solvents that includes analysis by HPLC, GC-MS, and ICP-MS. Extractables should be identified and their concentration estimated.
How can I find information on the toxicity of extractables found with my components? Evaluation of toxicology should be performed by a qualified toxicologist based on the dosage and route of administration. The BPSA has no specific recommendations regarding the toxicology of specific extractables or leachables. A PQRI document on extractables and leachables in OINDPs suggests an approach to address toxicology using LD50 with a 1,000× or 10,000× safety factor based on the dosage quantity (1). In addition, several structural activity relationship (SAR) databases are readily available to professional toxicologists. Examples include the “Carcinogenic Potency Database” (CPDB) (12) and the US Environmental Protection Agency’s “Distributed Structure-Searchable Toxicity Network” (DSSTOX) database (13). The PQRI document suggests a study design for addressing toxicity.
What are some classes of compounds the industry is highly concerned about as extractables? Various compounds can be of concern. Those that have been particularly identified include n-nitrosamines, polynuclear aromatics (sometime termed polyaromatic hydrocarbons, PAH), and 2-mercaptobenzothiozole, along with biologically active compounds such as bisphenol-A (BPA).
Do I need to validate my methodology for testing extractable analytes? For determination of leachables in products, it is currently industry standard to validate analytical methods according to ICH and USP criteria. This ensures appropriate levels of analytical precision and accuracy.
What substances, at what levels, can be extracted from a product? Various chemicals are added to plastic and elastomer formulations and can be extractables. They are typically proprietary to each manufacturer. These chemicals may be used as lubricants, plasticizers, antioxidants, stabilizers, antistatic agents, impact modifiers, catalysts, release agents, colorants, brighteners, bactericides, and blowing agents. Individual extractable compounds are too numerous to list, but examples include aromatic antioxidants such as butylated hydroxytoluene (BHT), oleamide, bromide, fluoride, chloride, oleic acid, erucamide, eicosane, and stearic acid.
The concentration of organic extractables in a drug product formulation depends on the composition of that formulation. Organic extractables will leach into formulations at a higher level if they have significant organic content or if compounds such as surfactants are present. The mass of an extractable that leaches is divided between all the doses in a production batch and thus is often significantly diluted from the worst-case extractable or leachable test results.
What is the time frame for developing extractables data using sponsor-requested liquids and test conditions? There are far too many manufacturing systems and formulations to suggest a specific timeline. Generally a product sponsor should anticipate four to six months to complete a controlled extraction study.
How are specific extractable analytes quantitated? Typically, extractable analytes are quantitated using chromatographic separations interfaced with precise detection systems. Examples of such analytical tools include HPLC equipped with diode-array detection, MS detection, or conductivity detection; GC equipped with MS detection or thermo energy analysis; and ICP or ICP-MS.
How are nonspecific analytes quantitated? The overall quantity of extractables or leachables can be estimated using nonspecific methods such as TOC and NVR analysis. Such nonspecific quantitation is especially useful in comparing materials before their final selection for a process.
Does the level of extractable analyte(s) need to be quantitated? If you’re going to use extractables data to qualify a material in a process, then you should determine the identification and concentration of the extractables. If you’re going to perform leachables studies in addition to obtaining extractable data, then it is not as important to quantitate the extractables. It is then important to quantitate identified leachables.
What kind of standard documentation should a vendor supply? It should be the goal of every vendor to understand the product it is providing to customers. However, every sponsor’s needs are different. It is the goal of this BPSA working group to provide guidance to product vendors on what and how much information should generally be available. Such guidance will be the focus of future BPSA committee papers.
How do extractables and leachables relate to shelf life or service life of components? It is well understood that plastic and/or elastomer materials have a limited shelf and service life. Addition of chemicals to plastic and elastomer formulations very often increase product shelf life and service life.