In 2004, the United States Food and Drug Administration (FDA) transferred regulation of many highly purified, “well-characterized” biopharmaceutical proteins from the Center for Biologics Evaluation and Research (CBER) to the Center for Drug Evaluation and Research (CDER), which until then had primarily regulated only synthetic, small-molecule drugs and chemical substances. The most novel/complex and the less-characterized biologics remained within CBER’s jurisdiction. This change complicated BPI’s mission somewhat. When the magazine was founded, we responded to questions from advertisers and public relations professionals about our coverage by pointing them to the CBER website. But suddenly, the answer wasn’t so easy.

Therapeutic products that transferred to CDER included monoclonal antibodies (MAbs); cytokines, growth factors, enzymes, immunomodulators, and thrombolytics; immunotherapeutics; and proteins (except clotting factors) extracted from microorganisms or animals, including recombinant versions of these products. Products remaining with CBER included vaccines; blood and related (e.g., fractionated) products; toxoids, toxins, antitoxins, antivenins, and venoms; gene therapies; products based on human or animal cells or parts thereof; allergen patch tests and allergenics; and some in vitro diagnostics. The “Approved Products” box lists US-marketed drugs that were directly affected by the switch at the time.
Part of our job as editors is to maintain distinctions among those product categories that BPI does and does not cover. It comes down to manufacturing technology: at the most basic level, the difference between synthetic and biological production — and downstream processing concerns that result from those differences. We are interested in biotherapeutics (whether peptide/protein-, gene-, or cell-based) and to a lesser extent vaccines/immunotherapeutics (especially the newer types made by cell culture), and biodiagnostics (particularly those based on recombinant proteins or related to and comarketed with biotherapeutics). In April 2010, Gary Hu did a good job of showing where the line is drawn for polypeptides (1). But my special report that fall and an article from Teva Pharmaceuticals in the issue alongside this anniversary supplement show how the line can get blurred (2, 3). Product Categories
A 2008 review article in BPI’s “Success Stories in Protein Expression” supplement reported that ~60 million patients worldwide had thus far benefited from biotherapeutics, estimating the 2004 market at US$33 billion and projecting $70 billion by the end of that decade (4). From 2003 to 2006, the author stated, regulators in Europe and the United States had approved 32 such products for human use, from hormones and growth factors to therapeutic enzymes, vaccines, and protein-based products. At the time, an additional 1,600 biopharmaceuticals were being evaluated in clinical trials — most concerned with immunological/autoimmune and oncological indications.
Monoclonal antibodies constitute by far the largest biotherapeutic product category, with the number of MAb candidates rising from 75 to 400 in the 2003–2006 period (4). Global MAb sales were $20.6 billion in 2006, with just 25 drugs on the market. Meanwhile, demand was ever increasing, making MAb-based products the most rapidly growing category of biopharmaceuticals.
When introduced as a product category (kicking off the biopharmaceutical industry in the late 1980s), MAbs were initially presented as “magic bullets” for targeting serious disease processes. Antibodies (immunoglobulins) are large Y-shaped proteins made by immune-system B-cells to identify and neutralize foreign invaders (e.g., viruses, bacteria). Part of the antibody structure binds to a unique part of a foreign antigen protein, tagging either the microbe or infected cell for attack by other parts of the immune system or neutralizing it directly. Naturally produced antibodies are polyclonal: Many different variations are made in response to a perceived attack.
Genetic engineering in the 1970s and 1980s led to production of monoclonal antibodies using hybridoma cells based on cancerous B cells known to produce only one antibody configuration. The specificity of these proteins to bind with their given targets like a lock and key made them exciting new drug prospects, but the nascent biotechnology industry faced unforeseen challenges in manufacturing and clinical testing. In 1986, the first FDA-approved MAb was a murine IgG2a CD3-specific transplant rejection drug called OKT3 (muromonab). But the industry didn’t see its second approval until 1994’s abciximab (ReoPro) for inhibition of glycoprotein IIb/IIIa in cardiovascular disease. Two more products were approved in 1997: daclizumab (Zenapax), a humanized IL-2Rα receptor (CD25-specific) also for transplant rejection, and rituximab (Rituxan/Mabthera), a chimeric CD20 for non-Hodgkin’s lymphoma.
That slow progress illustrates the problems that disappointed many early investors. The first MAbs were simple murine analogues with short in vivo half-lives (due to immunogenicity), limited tumor-site penetration, and inadequate host-effector functions. Chimeric and humanized antibodies have replaced murine antibodies in modern therapeutic antibody applications — as hybridoma technology was replaced by animal-cell culture production.
By the time BPI’s first issue was published in January 2003, there were 13 different MAb products on the US market. Now in 2012, the number has more than doubled — with hundreds more in development. The magic bullets are flying. In 2009, the first bispecific monoclonal antibody (BsMAb, BsAb) product was approved for marketing in the European Union: catumaxomab (Removab) is a rat–mouse hybrid MAb developed by Fresenius Biotech and Trion Pharma that binds to CD3 and EpCAM antigens for treating malignant ascites in patients with metastasizing cancer. Bispecific MAbs are fusion proteins that combine fragments of two different MAbs so they can bind to two specific antigens. For example, in cancer immunotherapy, BsMAbs can bind and bring together cytotoxic cells and tumor-cell targets for them to destroy.
Antibody–drug conjugates (ADCs) are not fusion proteins, which are genetically engineered to express in their combination form. Rather, ADCs are made by linking an expressed antibody (or fragment) to a payload drug (typically cytotoxic) as part of postproduction upstream processing (2). As in the BsMAb example above, the antibody portion of the ADC binds to a targeted cancer cell and is often internalized, where it releases a toxic payload drug. The specific MAb targeting lessens side effects associated with potent chemotherapies and radioactive isotopes. Hydrophilic linkers prevent these drugs from being rejected by cancer cells’ resistance mechanisms.
Companies involved in ADC development include Seattle Genetics, ImmunoGen Inc., and Spirogen Ltd. Like MAbs in the 1980s, conjugates are in their technological infancy, so those companies and others are working to solve problems related to manufaturing, safety, and efficacy. The first marketed ADC was gemtuzumab ozogamicin (Wyeth’s Mylotarg), approved in 2001 for acute myelogenous leukemia, but it has since been withdrawn due to poor phase IV trial results. Seattle Genetics’ brentuximab vedotin (Adcetris) was approved in 2011 for relapsed and refractory Hodgkin’s lymphoma and anaplastic large-cell lymphoma.
Nonantibody Proteins: Not all fusion and conjugate proteins are antibody-based, and they are just two classes of a growing number of other therapeutic proteins on the market and in development. These include blood and growth factors, complement proteins, enzymes and hormones, interleukins and other cytokines, receptor proteins, and subunit vaccines.
For example, PEGylation (conjugating a polyethylene glycol to a protein) is a popular conjugate technology used to increase the bioavailibility and half-life of products such as Pfizer’s pegvisomant (Somavert) recombinant somatropin approved for Pharmacia in 2003, Roche’s Mircera/CERA epoetin alpha approved in 2007, UCB’s certolizumab pegol (Cimzia) tumor necrosis factor approved in 2008 and marketed by Bayer and Schering, and Savient Pharmaceuticals’ Krystexxa (Puricase) pegloticase approved in 2010.
Remember that proteins participate in virtually every cellular process. There are probably millions of different configurations in the natural world, from enzymes that catalyze
biochemical reactions to muscle and cytoskeletal proteins that perform structural/mechanical functions and others involved in immune response, cell signaling, cell adhesion, and the cell cycle of division, growth, and death. Because these biomolecules are naturally occurring, they present fewer safety concerns than do synthetic chemicals when used as drugs. And because of their variety, they offer a never-ending array of therapeutic possibilities.
Vaccines: The vaccine industry has undergone a real renaissance since the turn of the century (5,6,7,8,9,10,11,12). Once considered old-fashioned technology — with seasonal influenza vaccines produced by inoculating hens’ eggs, for example — vaccines have come to the fore as immunotherapeutics as well as means of preventing infectious diseases. This is all attributable to advances in biotechnology that make it possible to manufacture such products using mammalian and insect cell culture. The range of vaccine categories has expanded from traditional live, attenuated, or killed whole organisms (e.g., the familiar polio and smallpox vaccines that all but eradicated both diseases in the 20th century) to include recombinant products such as Medimmune’s Flumist seasonal flu vaccine approved in 2003 and H1N1 swine-flu vaccine approved in 2009; tetanus, diptheria, and pertussis toxoids approved in 2005 for Chiron and GlaxoSmithKline; meningococcal conjugates approved in 2010 for Novartis; and subunit vaccines for human papillomavirus (HPV) approved for Merck in 2006 and GlaxoSmithKline in 2009.
Gene therapies are related to vaccines in that many in development use viruses as gene delivery vehicles — and thus involve similar production methods (13,14,15,16,17). But even though they’ve had millions of years to perfect the delivery of genes to cells, viruses aren’t the only tools for doing so with gene therapies. Plasmids and electroporation figure in as well (18, 19).
After tentative steps toward commercialization in the 1990s, gene therapy research in the United States was set back by the 1999 death of Jesse Gelsinger in a clinical trial significantly. The FDA suspended several other pending trials while reevaluating ethical and procedural practices in the field. Gene silencing through RNA interference emerged as an alternative in 2003. And patients were treated in small trials throughout the first decade of the 21st century. Still, neither the FDA nor the EMA has yet approved a human gene therapy product for sale even as research and development continues to grow in the United States and around the world. Treatments for cancer, cystic fibrosis, heart disease, hemophilia, wounds, infectious diseases, and graft-versus-host disease are all in the works.
Interestingly, China has emerged as a pioneer in some fields of gene therapy. In 2003, it became the first country to approve a product for market: an adenoviral-based gene therapy called Gendicine for squamous cell cancer. H-101 (Ocorine), an oncolytic virotherapy for head and neck cancers, became the second approved gene therapy in 2005. Commercialization of those products provided China with worldwide recognition in the field of gene therapy and led to some “medical tourism” — because neither has been offered to regulatory authorities in other countries.
Cell Therapies: Regenerative medicine appears to have surpassed gene therapy development elsewhere in the world (20, 21). Cell therapies have been approved for indications as seemingly trivial as facial wrinkles and as deadly serious as prostate cancer. And BPI has focused on the topic in two special issues so far (March 2011, March 2012). Developers of cell-based products are facing issues of scale and characterization for which biotherapeutics companies have already amassed some experience (22,23,24,25,26,27,28,29,30,31,32,33,34). We hope the younger industry will be able to learn from the more mature one (35,36,37,38,39,40,41).
Regenerative medicine — especially in the form of autologous cell and tissue therapies — is considered to be but one aspect of personalized medicine, which has been touted as “the next big thing” since the turn of the century.
Biodiagnostics: The 1998 approval of Genentech’s trastuzumab (Herceptin) breast cancer treatment — indicated only for patients who overexpress the HER2 gene — is often pointed to as the first in this new approach to therapeutics. Such targeted therapies go after aberrant molecular pathways in a subset of patients with a given cancer type.
Diagnostics are considered key to personalized medicine, and some are biotech in nature (e.g., MAb-based). BPI’s interested in so-called “biodiagnostics” is twofold: Some involve reagents with similar manufacturing processes as are used for the types of therapeutics we cover. And some support the use of those drugs in personalized medicine (42,43,44). In some cases, they might create a market for a product with previously disappointing sales (43). The Decade in Product Approvals
Information for regulatory approvals of biopharmaceutical products is readily available online, especially through BPI author Ron Rader’s comprehensive database (www.biopharma.com). In 2007, he wrote a detailed two-part article “concerning generic biopharmaceutical paradigms, terminology, and nomenclature issues: how we think of, define, name, and regulate these products” (45, 46). In reviewing the background, providing perspective, and discussing nomenclature, legislation, and public controversies, he also managed to quite neatly define BPI’s own product coverage niche, as described herein. And his contribution to our highly successful “30 Years of Product Approvals” wallchart was invaluable; we could not have done it without him.
(Tables 12345678910) list US biopharmaceutical products that the FDA has approved during BPI’s first 10 years — up through March 2012, that is. All the information came from Rader’s database (47), and I am grateful for his making that available.Table 1: US product approvals 2003 (49)
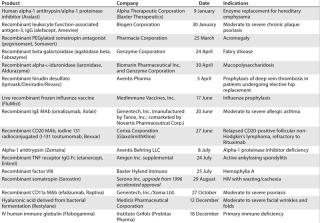
Table 2: US product approvals 2004 (49)
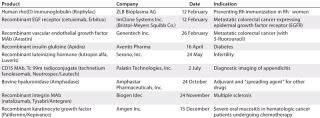
Table 3: US product approvals 2005 (49)
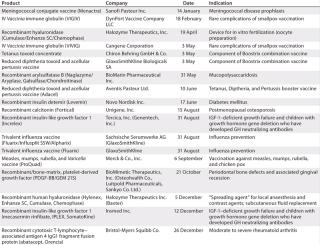
Table 4: US product approvals 2006 (49)
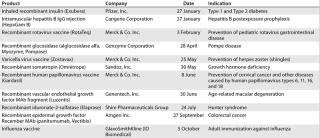
Table 5: US product approvals 2007 (49)

Table 6: US product approvals 2008 (49)
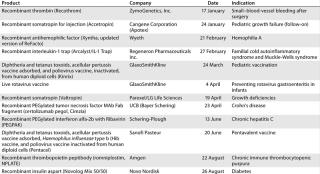
Table 7: US product approvals 2009 (49)
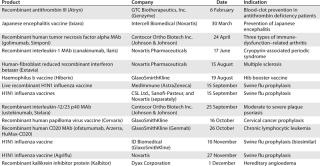
Table 8: US product approvals 2010 (49)

Table 9: US product approvals 2011 (49)

Table 10: US product approvals through March 2012 (49)

Author Details
Cheryl Scott is cofounder and has been senior technical editor of BioProcess International since the first issue.
REFERENCES
1.) Hu, G 2010. Understanding the Basics of Peptide and Protein Production. BioProcess Int. 8:22-25.
2.) Scott, C. 2010. Protein Conjugates: Facing the Challenges of Their Development and Manufacture. BioProcess Int. 8:28-37.
3.) Weinstein, V. 2012. Looking at the Recent FDA Biosimilar Guidelines: Immunogenicity Concerns and Extension to Other Classes of Drugs. BioProcess Int. 10:in press.
4.) Coco-Martin, JM, and MM. Harmsen. 2008. A Review of Therapeutic Protein Expression By Mammalian Cells. BioProcess Int. 6:S28-S33.
5.) Scott, C. 2004. Classifying Vaccines: From Cowpox to the Cutting Edge. BioProcess Int. 2:S14-S23.
6.) Gerson, DF, and B. Mukherjee. 2005. Manufacturing Process Development for High-Volume, Low-Cost Vaccines. BioProcess Int. 3:42-47.
7.) Mørtz, E. 2008. Proteomics Technology Applied to Upstream and Downstream Process Development of a Protein Vaccine. BioProcess Int. 6:36-43.
8.) Kowolenko, MD. 2009. The Vaccine Renaissance. BioProcess Int. 7:72.
9.) Ball, P, C Brown, and K. Lindström. 2009. 21st Century Vaccine Manufacturing. BioProcess Int. 7:18-28.
10.) McLeod, LD. 2009. Novel Vaccines and Virology. BioProcess Int. 7:S34-S39.
11.) Whitford, WG. 2010. Using Disposables in Cell-Culture–Based Vaccine Production. BioProcess Int. 8:S20-S27.
12.) Scott, C. 2010. Speeding Vaccine Development and Production. BioProcess Int. 8:S36-S42.
13.) Gombold, J. 2006. Lot Release and Characterization Testing of Live-Virus–Based Vaccines and Gene Therapy Products, Part 1: Factors Influencing Assay Choices. BioProcess Int. 4:46-54.
14.) Gombold, J. 2006. Lot Release and Characterization Testing of Live-Virus–Based Vaccines and Gene Therapy Products, Part 2: Case Studies and Discussion. BioProcess Int. 4:56-65.
15.) Hannaman, D, R Bernard, and M. de las Alas. 2008. Delivery Technology Reenergizes DNA Drug Development. BioProcess Int. 6:44-52.
16.) Filipe, V, W Jiskoot, and A. Hawe. 2011. Understanding Virus Preparations Using Nanoscale Particle Characterization. BioProcess Int. 9:44-51.
17.) Ausubel, LJ. 2012. Production of CGMP-Grade Lentiviral Vectors. BioProcess Int. 10:32-43.
18.) Hitchcock, AG. 2010. Scale-Up of a Plasmid DNA Purification Process. BioProcess Int. 8:46-54.
19.) Kim, JJ. 2011. Electroporation-Enhanced Delivery of DNA Vaccines. BioProcess Int. 9:S10-S14.
20.) Sofer, G. 2005. A European PTC Document on Xenogeneic Cell Therapies. BioProcess Int. 65 3:18-20.
21.) Knocke, C, and J. Vogt. 2008. Taking the Next Step: Dynamic Cultivation Opens Commercial Use for Stem Cells. BioProcess Int. 6:68-73.
22.) Hampson, B, J Rowley, and N. Venturi. 2008. Manufacturing Patient-Specific Cell Therapy Products. BioProcess Int. 6:60-72.
23.) Fitzpatrick, I. 2008. Cellular Therapy Success Through Integrated Automation. BioProcess Int. 6:S32-S37.
24.) Benz, K, and J. Mollenhauer. 2008. Quality Assessment in 3D Cultures of Disc-Chondrocytes. BioProcess Int. 6:54-59.
25.) McIntyre, CA, BT Flyg, and TC. Fong. 2010. Fluorescence-Activated Cell Sorting for CGMP Processing of Therapeutic Cells. BioProcess Int. 8:44-53.
26.) James, D. 2011. Therapies of Tomorrow Require More Than Factories from the Past. BioProcess Int. 9:S4-S11.
27.) Shaw, R. 2011. Stem-Cell–Based Therapies. BioProcess Int. 9:S20-S25.
28.) Monesmith, TT. 2011. Meeting the Challenges in Manufacturing Autologous Cellular Therapies. BioProcess Int. 9:S38-S41.
29.) Brandenberger, R. 2011. Cell Therapy Bioprocessing. BioProcess Int. 9:S30-S37.
30.) Rios, M. 2011. Technologies on the Cutting Edge. BioProcess Int. 9:S26-S29.
31.) Buckler, RL. 2011. Opportunities in Regenerative Medicine. BioProcess Int. 9:S14-S19.
32.) Scott, C. 2011. An Update on Cell-Based Technologies. BioProcess Int. 9:32-50.
33.) Rowley, J. 2012. Meeting Lot-Size Challenges of Manufacturing Adherent Cells for Therapy. BioProcess Int. 10:S16-S22.
34.) James, D. 2012. Small–Batch-Size Production. BioProcess Int. 10:S30-S35.
35.) Rios, M. 2011. Industry Roundtable. BioProcess Int. 9:S42-S44.
36.) Rios, M. 2011. Successful Commercialization Through Industry Collaboration. BioProcess Int. 9:S12-S13.
37.) Prieels, J-P. 2012. Mastering Industrialization of Cell Therapy Products. BioProcess Int. 10:S12-S15.
38.) Davie, NL. 2012. Streamlining Cell Therapy Manufacture. BioProcess Int. 10:S24-29.
39.) Pritchett, T, and L. Little. 2012. Hard Cell. BioProcess Int. 10:S36-S48.
40.) Rosenblatt, B. 2012. Meeting Regulatory Challenges for Cell-Based Therapies. BioProcess Int. 10:S8-S11.
41.) Jones, S, SD McKee, and HL. Levine. 2012. Emerging Challenges in Cell Therapy Manufacturing. BioProcess Int. 10:S4-S7.
42.) Fuller, A. 2008. Will Diagnostics Return to Its Roots?. BioProcess Int. 6:80.
43.) Levesque, P. 2009. Overcoming Barriers to Success. BioProcess Int. 7:20-22.
44.) Tinsley, R. 2011. Questions to Consider When Thinking About Companion Tests. BioProcess Int. 9:10-13.
45.) Rader, RA. 2007. Biopharmaceutical? Biogeneric? Follow-On Protein? Biosimilar? Follow-On Biologic? Part 1: Introduction and Basic Paradigms. BioProcess Int. 5:28-38.
46.) Rader, RA. 2007. What Is a Generic Biopharmaceutical? Biogeneric? Follow-on Protein? Biosimilar? Follow-on Biologic? Part 2: Information, Nomenclature, Perceptions, and the Market. BioProcess Int. 5:20-28.
47.) Rader, RA. 2012. Recent US Approvals. BIOPHARMA: Biopharmaceutical Products in the US and European Markets