Biologic drugs are subject to unique regulatory and technical requirements because of their origin and expression in genetically engineered host cells, as well as their underlying physicochemical properties and elaborate purification processes. One such requirement is the accurate monitoring and effective removal of process-derived impurities such as host-cell proteins (HCPs) and DNA/RNA, viruses, cell culture media, chromatographic leachates, and so on (1). Of those impurities, HCPs are perhaps the most challenging to accurately monitor. Each expression system’s proteome consists of thousands of different proteins, some of which can copurify with a biological drug. So the HCP monitoring method must be a multianalyte assay that can detect a great majority of the protein impurities that could be present in a batch of drug substance (2, 3).

One of the most widely used methods for monitoring HCPs is the enzyme-linked immunosorbent assay (ELISA) due to its high sensitivity (low ppm) and potential for broad specificity for the population of HCPs (1,2,3,4). An HCP ELISA uses a polyclonal “antibody reagent” (anti-HCP antibodies) grown in animals against HCP preparations from a product expression system. The accuracy of this immunoassay depends on the antibody reagent’s ability to detect essentially all potential HCP impurities. So that reagent must be validated to be suitably specific for its intended use by testing for the percentage of immunodetection it offers for the total population of HCPs before it is used in the HCP ELISA.
PRODUCT FOCUS: RECOMBINANT PROTEINS
PROCESS FOCUS: PRODUCTION
WHO SHOULD READ: ANALYTICAl, QA/QC, PROCESS DEVELOPMENT
KEYWORDS: HOST-CELL PROTEINS, ASSAY DEVELOPMENT, REPLICATES, QUALIFICATION
LEVEL: INTERMEDIATE
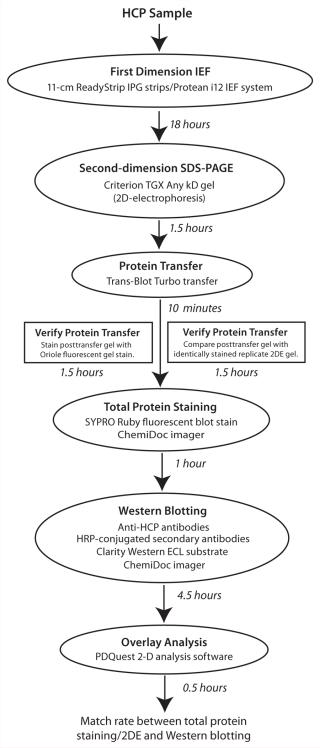
Among multiple technical approaches for evaluating the antibody reagent, a combination of two-dimensional electrophoresis (2DE) and Western blotting (WB) is the current “gold standard” for visualizing total HCP populations and assessing the percent immunocoverage of an anti-HCP polyclonal antibody reagent (5, 6). The standard workflow for this evaluation comprises four steps. First, 2DE of host-cell protein preparations is performed in replicate gels. Second, one post-2DE gel is processed for total protein detection by a sensitive protein stain. Third, proteins from replicate 2DE gels are transferred to a solid support membrane such as nitrocellulose or polyvinylidene fluoride (PVDF) for WB, with the polyclonal anti-HCP antibodies being evaluated. Finally, images from 2DE and WB membranes are overlaid to obtain a match rate for determining the percentage of HCPs that the anti-HCP antibodies can detect. Although it is widely used, the laborious nature of this workflow — and experimental variation inherent to comparisons between replicate 2DE analyses — can affect overall reliability of final results. That complicates downstream decisions for effective HCP impurity profiling of purified biologics, including identification of potential protein impurities “missed” by anti-HCP antibodies and development of orthogonal methods to monitor those impurities.
To increase reliability of anti-HCP antibody evaluations and facilitate downstream decisions for effective HCP impurity profiling, we present here enhancements to the standard 2DE and WB workflow. Further, we apply these enhancements as a model example to the evaluation of a commercially available anti-HCP antibody reagent.
Materials and MethodsWe purchased the following materials from Bio-Rad Laboratories: a ReadyPrep 2D cleanup kit and ReadyPrep rehydration/sample buffer, ReadyStrip immobilized pH gradient (IPG) strips, Criterion gels, Trans-Blot Turbo PVDF transfer packs, Bio-Safe Coomassie stain, Oriole fluorescent gel stain, SYPRO Ruby protein blot stain, blotting grade-blocker, and Clarity Western enhanced chemiluminescence (ECL) substrate. NuPage gels and Gibco cell-culture media were purchased from Life Technologies (now Thermo Scientific), and ultrafiltration protein concentration/buffer exchange cartridges were purchased from Sartorius. And we used anti-HCP antibodies to Chinese hamster ovary (CHO) proteins purchased from Cygnus Technologies (3G-0016-PA) as the primary antibody and HRP-conjugated bovine antigoat antibody purchased from Santa Cruz Biotechnology as the secondary antibody.
Sample Preparation: Frozen rat livers from Pel-Freez Biological were extracted in a solution consisting of 8M Urea, 4% CHAPS electrophoresis reagent, 40mM dithiothreitol (DTT), and 0.2% Bio-Lyte ampholyte 3–10. Following that extraction, we centrifuged the sample at 14,000g for 15 minutes and then collected the supernatant for 2DE. We used these samples for determining the influence of gel chemistry on transfer efficiency across both isoelectric point (pI) and molecular weight (MW) using the Trans-Blot Turbo system.
Adherent cultures of CHO-K1 cells were adapted to a medium consisting of 95% Gibco protein-free defined medium from Life Technologies and 5% Ham’s medium supplemented with 10% phosphate-buffered saline (PBS). We processed cells for collection of total protein (by sonication in 7M urea/2M thiourea and clarification by centrifugation) or secreted protein (by transferring them to 100% protein-free defined medium). Secreted protein was prepared by 100-fold centrifugal concentration (10,000 MWCO), then treated with the ReadyPrep 2-D cleanup kit and resuspended in 8M urea. We used CHO-K1–derived samples for 2DE and WB workflow development and evaluation of anti-CHO HCP antibodies.
2DE, Protein Transfer, Total Protein Staining, and WB: For rat-liver samples, we carried out first-dimension separation using 11-cm pH 5–8 ReadyStrip IPG strips under standard isoelectric focusing (IEF) conditions. Second-dimension separations involved Novex Bis-Tris (4–12)%, Novex Tris-Glycine (4–12)%, Criterion TGX Any kD, or Criterion Tris-HCl (8–16)% gels transferred to PVDF membranes using the Trans-Blot Turbo system. Following protein transfer, we stained the blots with SYPRO Ruby blot stain and visualized them using a VersaDoc 4000 imaging system from Bio-Rad. To visualize protein remaining in the 2D gels (not transferred), we stained those gels with Biosafe Coomassie stain and scanned them on a Molecular Imager GS-800 calibrated densitometer from Bio-Rad.
For CHO-K1 cell-derived samples, we carried out the first-dimension separation using the Protean i12 IEF cell in “gel-side-up” mode with 150 µg total protein on an 11-cm ReadyStrip pH 3-10 NL strips. The CHO-secreted sample solution was 8M urea, 4% CHAPS, 40mM DTT, and 0.2% Bio-Lyte 3/10 carrier ampholyte; for CHO cell lysate, the 8M urea was replaced with 7M urea and 2M thiourea. We carried out second-dimension separations using Criterion TGX Any kD gels.
After 2DE of CHO-K1 samples, we stained the gels either with stained with Oriole fluorescent gel stain or transferred them
to PVDF using the Trans-Blot Turbo system. Following transfer, the gels were stained with Oriole fluorescent gel stain (to visualize remaining protein). We stained the blots with SYPRO Ruby stain (for total protein) and imaged them. Subsequently, they were directly processed for blocking with 5% blotting-grade blocker — nonfat dry milk in Tris-buffered saline supplemented with 0.05% Tween-20 (TTBS) — followed by immunodetection under optimized concentrations of anti-HCP antibodies (primary antibodies diluted 1:150 in TTBS) and HRP-conjugated secondary antibodies (diluted 1:3,000 in TTBS). After washing them multiple times (six times for five minutes each), we used Clarity Western ECL substrate for chemiluminescent development of Western blots.
Image Capture and Overlay Analysis: We used a ChemiDoc MP imager to visualize CHO-K1 samples processed by 2DE (fluorescence images of stained gels and blots) and WB (chemiluminescent images). For image overlay analysis or spot counting, we used PDQuest 2D analysis software. It generated a “master gel” that included all spots detected on both 2DE and WB analyses and expressed the “match rate” as a percentage determined using
Equation 1: Formula for determining match rate, expressed as a percentage of spots detected on the “master gel”
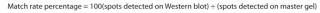
Factors Affecting Reliability of Standard 2DE and WB Workflow: In designing workflow improvements for 2DE and WB for anti-HCP antibody evaluations, we initially determined the potential for error in the standard workflow. Replicate 2DE gels of HCP preparations are processed for two analyses performed in parallel: total HCP staining on one 2DE gel and WB (with anti-HCP antibodies) of the HCPs transferred from a replicate 2DE gel to a solid support membrane. Images from both analyses are overlaid to obtain a match rate (the percentage of HCPs detectable by anti-HCP antibodies). However, if the two replicate 2DE analyses differ significantly or protein transfers after 2DE to the WB membrane are inefficient, then erroneous match-rate estimates would result. Each of those factors (variability between replicates and protein transfer efficiency) thus affects reliability of final results obtained using the standard 2DE–WB workflow.
To test for the extent of potential variability between replicate 2DE gels, we initially performed three replicate 2DE analyses of identical amounts of the same quantity (150 µg) of CHO-cell–secreted protein and processed them further (Figure 1, Table 1). After 2DE, we stained the gels with Oriole fluorescent stain similarly and processed them for spot counting and matching with the PDQuest software under identical settings. As Figure 1, LEFT and Table 1 show, the replicates appear qualitatively similar, and their spot numbers are roughly comparable (435, 449, and 454 spots, respectively).


To determine more rigorously the replicate variability, we generated a master gel (a virtual reference gel containing all detected spots from the three replicates) and performed overlay analysis between it and each of replicate (Figure 1, RIGHT). For consistency, we analyzed all images under identical settings with no manual spot editing. If the 2DE replicates were nearly identical to one another — as suggested by the relatively similar number of detectable spots from each one — then we reasoned that an overlay with the master gel should result in a near-100% match rate.
However, as Figure 1, RIGHT and Table 1 show, a significant number of detected spots on each replicate are unmatched to the master gel — resulting in 70%, 72%, and 73% match rates for each comparison, respectively. Further, the number of spots common to all three 2DE replicate analyses (289) is even lower (Table 1), pointing to significant variability between replicate 2DE analyses. These results highlight significant error potential in match-rate determinations from the standard workflow wherein replicate 2DE analyses are processed for total protein staining and WB. Thus, to minimize differences in spot counts from gel to gel, two different 2D gels should not be used even if they are replicates. For reliable final results, it would be best to perform total protein staining after 2DE and WB on the same membrane — after ensuring efficient protein transfers from the 2DE gel.
Efficiency of protein transfer following 2DE analysis is another important consideration. Protein transfer efficiency can be influenced by sodium-dodecyl sulfate– polyacrylamide gel electrophoresis (SDS-PAGE) chemistry as well as the transfer system/method used (e.g., tank-transfer system or the much faster semidry transfer/Trans-Blot Turbo systems and associated buffers). The speed of semidry transfer systems makes them an attractive option, but it is critical that they do not compromise transfer efficiencies across the entire range of pI and MW. Further, this evaluation needs to be performed in the context of potential SDS-PAGE chemistry influences. So we evaluated the influence of four different SDS-PAGE chemistries on transfer efficiencies using the Trans-Blot Turbo system. Given the limited amount of CHO-derived material in our possession, we used rat-liver lysate as a model complex mixture of proteins with a wide pI and MW range for these experiments.
Identical quantities of rat-liver lysate were resolved in parallel by isoelectric focusing (first dimension) using pH 5–8 IPG strips (Figure 2). Subsequently, each was resolved in the second dimension using one of the four SDS-PAGE chemistries under evaluation: Novex Bis-Tris (4–12)%, Novex Tris-Glycine (4–12)%, Criterion TGX Any kD, and Criterion T
ris-HCl (8–16)% gels. After performing 2DE, we transferred the proteins to PVDF membranes using the Trans-Blot Turbo system for seven minutes at 25V/2.5A. To visualize the transferred proteins, we stained the membranes using SYPRO Ruby; to visualize proteins remaining on each gel (not transferred), we stained the gels with Bio-Safe Coomassie Blue. As Figure 2 shows, the post-transfer Criterion TGX gel has essentially no Coomassie-reactive spots, indicating highly efficient protein transfer efficiency across pI and molecular weight compared with the other gel chemistries.
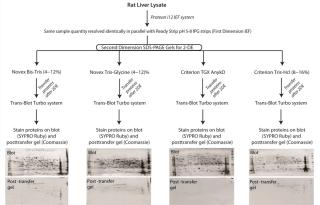
Those results demonstrate that for reliability of final results, it is desirable to perform post-2DE total protein detection and WB analyses on the same membrane. Additionally, they describe an optimal combination of SDS-PAGE gel chemistry and protein transfer system for highly efficient protein transfer post-2DE for downstream same-membrane analyses.
Compatible Options for Same-Membrane 2DE and WB Analyses: Next, we considered two options that might be compatible with same-membrane 2DE total protein detection and subsequent WB (7):
-
minimal fluorescent-dye labeling (e.g., charge-neutral Quasar/Cye dyes or equivalent) of HCP mixtures to be followed by chemiluminescent or fluorescent WB
-
sensitive, noncovalent protein staining (e.g., SYPRO Ruby fluorescent stain) that is reversible and thus compatible with WB applications.
The first option above — minimal fluorescent dye labeling — has the potential for ~5× higher sensitivity over the second option; further, it is more sensitive than silver-stained total protein detection on gels (8). However, the former option also creates two populations of protein mixtures: those that are dye-labeled (and covalently modified, resulting in slight MW or pI changes to the proteins) and those that are unlabeled (and thus unmodified). Such a twofold increase in protein complexity effectively reduces the overall resolution of 2DE gels by about a factor of two, prompting use of larger-format 2DE gels. That extends the overall 2DE and WB workflow to ~4–5 days, requiring extensive optimization for efficient protein transfer, and necessitates more careful manual handling. In addition, covalent modification of proteins (by fluorescent dye labeling) can affect their immunogenicity toward the anti-HCP antibodies, potentially influencing accuracy of the experimentally determined match rates.
Given those, we preferred the second option: noncovalent, reversible staining of HCPs transferred to PVDF membranes using SYPRO Ruby stain, followed by WB with anti-HCP antibodies. We reasoned that the expected lower sensitivity of SYPRO Ruby stain could be overcome by using a higher amount of the HCP mixture to begin with (about a fivefold increase over that used with the minimal fluorescent dye-labeling option, ~150 µg protein amount for 11-cm IPG strips). Also, because this option does not effectively decrease 2DE resolution, we could use medium-sized 2DE gel configurations optimized for fast, efficient protein transfer (11-cm IPG strips, Criterion TGX Any kD gels, and the Trans-Blot Turbo system) (Figure 2), with a streamlined workflow (about two days total). Finally, SYPRO Ruby stain is reversible and does not covalently modify proteins, unlike dye-labeling (7). So it would not affect immunogenicity of the proteins being analyzed — thus enabling accurate match-rate determinations.
Application of Same-Membrane 2DE and WB Analyses for Evaluations of Anti-CHO HCP Antibodies: Next, we implemented our new 2DE–WB workflow (Figure 3) for evaluating a commercially available anti-CHO antibody reagent against two CHO-derived samples (CHO total protein and secreted protein). Briefly, we performed a single 2DE analysis of each sample using optimized conditions for the first-dimension IEF (11-cm pH 3–10 NL ReadyStrip IPG strips) and 2DE (Criterion TGX Any kD gels). Afterward, proteins were transferred to PVDF membranes in under 10 minutes using the Trans-Blot Turbo. To monitor transfer efficiency, we stained the post-transfer gel with Oriole fluorescent gel stain (9) and compared it with a replicate 2DE gel of each sample stained identically. Transferred proteins were visualized using SYPRO Ruby fluorescent protein staining and subsequently processed for chemiluminescent WB with anti-CHO antibodies and HRP-conjugated secondary antibodies. After imaging on the multipurpose ChemiDoc MP imager, we performed spot matching and image alignment using PDQuest 2D analysis software. Figure 4 shows images from each step within this workflow for CHO total-protein (Figure 4, TOP) and CHO secreted-protein (Figure 4, BOTTOM) samples.

Here are the key benefits of our workflow over the standard 2DE–WB procedure used for anti-HCP antibody evaluations:
-
Same-membrane analysis not only resolves reproducibility issues between 2DE replicates, but it also enables high-confidence spot matching and image alignment using 2D analysis software
-
The fast (<10 minutes), highly efficient protein transfer leaves no detectable protein left behind (Figure 4; comparing Oriole fluorescent gel stained replicate 2DE gel with the posttransfer gel) increases thoroughness of downstream analyses
-
Fluorescent noncovalent protein staining and chemiluminescent WB analyses achieves high-sensitivity protein detection without loss of 2DE resolution or potentially erroneous match-rate estimates (due to modified immunogenicities of HCPs, which may be seen with covalent labeling of fluorescent dyes)
-
Using the same imager for both analyses generates directly comparable high-resolution images
-
The enti
re workflow generates high-confidence results in under two days.
As Figure 5 shows, a 47% match rate was obtained using the anti-CHO HCP antibodies against CHO secreted protein and 20% for total protein using the formula in

The higher match rate seen with CHO secreted-protein samples than with CHO total-protein samples can be expected because antibodies generated by the commercial vendor were against proteins released into growth medium. Using a more similar sample to that used by the vendor for antibody generation — or those that are immunopurified — could further increase the match rate. We chose not to do that here because our primary focus was to develop needed improvements for reliable evaluations of anti-HCP antibodies. Performing such evaluations with speed and confidence is important in enabling downstream decisions regarding how best to monitor potential protein impurities missed by anti-HCP antibodies/HCP ELISAs during impurity analyses of biologics.
Streamlining for Speed and ReliabilityWe present here a streamlined 2DE and WB workflow for highly reliable evaluations of anti-HCP antibodies that can be completed in under two days. The enhanced reliability and speed achieved in this workflow stems from eliminating replicate 2DE analyses, using medium-sized IPG strips and SDS-PAGE gels, quickly and efficiently transferring protein after 2DE, and performing highly sensitive (fluorescent) total protein staining and chemiluminescent WB on the same membrane for reliable spot matching. Because the proteins remain unmodified chemically (unlike with alternatives such as fluorescent dye-labeling), no immunogenicity of proteins toward the anti-HCP antibodies is lost in the process, nor is there any loss in sample resolution usually associated with minimal covalent labeling strategies. The latter facilitates use of medium-sized 2DE gels that further accelerate the entire workflow. As proof of concept, we used this new workflow to reliably evaluate commercially available anti-CHO HCP antibodies against CHO total-protein and secreted-protein samples.
Author Details
Corresponding author Tom Berkelman and Adriana Harbers are senior staff scientists in the laboratory sciences division, and Sricharan Bandhakavi is laboratory separations global product manager at Bio-Rad Laboratories, Hercules, CA 94547. ChengJun Sun is a research scientist at The First Institute of Oceanography in China.