In cancer treatment, early diagnosis and targeted therapies are assumed to yield the highest cure rates. However, most current methods are limited by their low sensitivity to early disease and a lack of specificity for targeted cell killing. Newly developed, activatable immunoconjugates assist in the accurate detection of cancer through in vivo imaging with high target-to-background contrast (1,2). They also provide for the possibility of highly specific, light-mediated treatment with minimal effects on healthy cells surrounding tumors (3). In fact, the optical strategies for designing highly targeted and specific diagnostic and therapeutic technologies are identical. The major difference in the two approaches is the form in which the emitted energy is released: as light for diagnostic purposes and as heat and/or oxidation for therapeutic purposes. Here I discuss strategies for designing activatable immunoconjugates for use in both diagnosis and treatment of cancer.
PRODUCT FOCUS: A
PROCESS FOCUS: PRODUCT DESIGN AND PRECLINICAL TESTING
WHO SHOULD READ: ANALYTICAL AND PRODUCT DEVELOPMENT
KEYWORDS: DIAGNOSTICS, CONJUGATION, LIGHT DETECTION, PROBES, DYES, LABELING, DETECTION, PRECLINICAL TESTING
LEVEL: ADVANCED
Conventional cancer diagnostic imaging methods using contrast agents — such as angiography, computed tomography, and magnetic resonance imaging (MRI) — have had a profound impact on cancer treatment. But improvements are needed in both sensitivity and specificity of such approaches.
Radionuclide (isotope) imaging is a good example illustrating the current limitations. Although it provides physiologic information (regarding blood flow, perfusion, and/or lymphatic flow) or metabolic information (regarding phosphates, sugars, nucleic acids, and some amino acids), radionuclide imaging does so with low spatial resolution and insufficient specificity for diagnosing cancers. Only a few molecularly targeted radioactive probes (e.g., somatostatin analog and radiolabeled antibodies) have been approved for use in clinical practice. Because such isotopes constantly emit radiation as a signal, they are termed “always-on” agents. Target-to-background signal ratios (TBRs), which determine sensitivity, rely on the in vivo biodistribution of radionuclides regardless of their proximity or interaction with target molecules or tissues. Unbound circulating probes and their catabolites produce a nonspecific background signal, so prolonged circulatory times decrease TBR and compromise cancer detection.
Molecular imaging, which is seen as an important advance in imaging sciences, offers the possibility of obtaining in vivo, target-specific information with high sensitivity and specificity. To achieve this goal, it may be necessary to design superior probes that can achieve increased TBRs. Designing such probes requires either maximizing signal from targets or minimizing signal from the background, or both. Doing so leads to improved TBR, which in turn improves sensitivity and specificity for detecting diseases by such imaging. A long-term approach to maximizing target signals is based on altering biodistribution and pharmacokinetics of imaging probes. However, recent advances in activatable agents that emit a signal only after enzymatic digestion or acidification represent an exciting new strategy for improving TBRs.
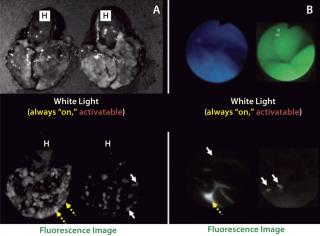
In contrast with radionuclide imaging, optical imaging using fluorescent probes is a relatively new capability in medicine. It offers advantages of lower costs, portability, and real-time capabilities. Of course, the limited depth of light penetration in living tissue makes it necessary to reserve fluorescence optical imaging for surfaces such as those encountered during endoscopic or surgical procedures. A unique feature of fluorescence is that optical probes can be designed that do not emit light if they are unbound, but that generate light upon enzyme catabolism or lysosome internalization. This attribute of “turning on” only when interacting or binding to a target molecule fits the definition of an “activatable” or “smart” imaging probe. Such agents have little or no background signal; they generate signal only after being bound to a specific molecular target (1,4). Activatable fluorescence probes maximize target signals while minimizing background signals, which results in higher TBRs than can be achieved by conventional “always-on” imaging agents (Figure 1).
Once we had developed a number of methods for designing activatable fluorescence-imaging probes (1), we attempted to design imaging agents that would be feasible for translation into humans. Below are two examples of such agents that are currently in development for clinical application.
For Prostate Cancer: The first is an activatable fluorescent imaging probe for prostate-specific membrane antigen (PSMA) made up of a humanized anti-PSMA antibody (Ab) or fragment conjugated to indocyanine green (ICG) (5,6). A similar humanized anti-PSMA Ab (HuJ591) has shown good safety profiles in therapeutic and diagnostic clinical trials for prostate cancer patients. ICG has been safely used by millions of patients either as a liver functional agent or for ophthalmologic retinal examinations for over 50 years, being injected at >1,000 times the dose used in this Ab-conjugation. Therefore, although we still need to ensure the safety of our resulting Ab-ICG conjugates, both major components of each conjugate have been used safely in humans already.
To successfully synthesize human doses of compounds with high yield, we refined the conjugation chemistry (7,8). To stably and massively synthesize clinical grade of conjugates but still retain preferable activatable characteristics, a short polyethylene glycol (PEG) spacer at the conjugation site helps us obtain monodispersed final products with superior yield. These efforts have enabled us to move the Ab-ICG conjugates on toward clinical trials.
A Surgical Application: The second example of a translatable, activatable optical probe is a small-endopeptidase enzyme for assisting surgery or endoscopic procedures when topically sprayed onto epithelial membranes. Topical use minimizes the dose from what would be required for systemic intravenous injection, and this lower dose lessens chances for adverse events. Although enzyme expression is not as specific as targeting tumor membrane markers (e.g., growth factor receptors for targeting cancers), enzymes related to cell proliferation or invasion can cover greater numbers of cancers than tumor membrane markers can. That makes enzymes more economically viable as a potential product.
For practical use during surgical or endoscopic procedures, a useful imaging agent should not contaminate “background” normal tissue, but it should
be activated within minutes after spraying to regions of interests. While taking that and other issues into account, we designed a gamma-glutamyltransferase (gGGT) activatable imaging probe based on hydroxymethyl rhodamine using the spirocyclic quenching/activation mechanism immediately after cleavage by gGGT. This gGGT-activatable probe can make out tiny tumor nodules that arise from disseminated peritoneal ovarian cancer (Figure 2) or in colitis-associated colon cancer. They become optically visible within a minute of exposure (9,10). This agent currently is used ex vivo on surgical specimens for selecting useful cancers in clinical studies.

Imaging can play a major role in diagnosis, but it also can be used to guide and deliver therapeutics directly. From a physics perspective, excitation energy can be dissipated in the form of fluorescent light for diagnosis. Alternatively, it can be exchanged for heat or oxidation in cytotoxic treatment (Figure 3).
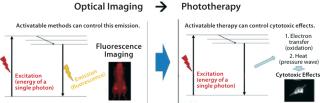
A distinction must be made between the established photodynamic therapy (PDT) technique and this newly developed photoimmunotherapy (PIT). The former has been in limited clinical use for several decades. Photofrin (porfimer) is the PDT agent in most common use, having been used to treat endobronchial, esophageal, and bladder cancers. This and other related compounds are highly hydrophobic photosensitizers that are internalized in cells and generate reactive oxygen species when they are exposed to light in the presence of oxygen. Those molecular species are typically toxic to cells and cause cell death.
Although there are a few clinical uses of PDT, its widespread adoption has been limited because the current compounds distribute somewhat nonspecifically in vivo and permeate into normal cells, which causes off-target toxicities. Specifically, because of slow clearance rates (a 23-day half-life) and nonspecific uptake, patients must often refrain from being exposed to sunlight for weeks to months after exposure to PDT agents to avoid skin injury. General use of PDT thus is limited by its potential side effects. Some companies have attempted to targeted PDT by conjugating photosensitizers to targeting moieties such as antibodies, but delivery issues have arisen related to the large size of the resulting conjugates and hydrophobic nature of photosensitizers. Those limit their achievable concentrations.
While screening for phototoxic agents that could be conjugatable to antibodies without changing their pharmacokinetics, we found antibody-photosensitizer conjugates that were highly cell specific with no off-target effects and largely independent of the need for internalization. Specifically, by conjugating a phthalocyanine dye (IR700) to trastuzumab — the anti- HER2 monoclonal antibody (MAb) — or panitumumab (anti-EGFR, anti-HER1), it was possible to kill cancers with the application of relatively modest amounts of near-infrared (NIR) light supplied by a light-emitting diode (LED). The required light levels did not even raise skin temperature. Interestingly, both resulting conjugates were ineffective against cells that did not express HER2 or EGFR/HER1, respectively. Thus, we achieved excellent tumor responses without normal tissue toxicity. We called this promising method (as illustrated in Figure 4) photoimmunotherapy (PIT) (3).
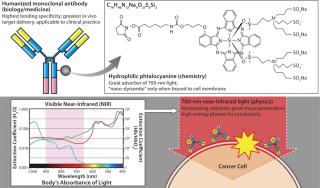
IR700 is a hydrophilic phthalocyanine dye that acts as a photosensitizer when excited with NIR light. It is readily and covalently conjugated to any MAb, including FDA-approved humanized MAbs that target human epidermal growth factor receptor (EGFR) type 1 (HER1) and 2 (HER2) among many others. When exposed to light, the resulting conjugate induces highly selective cell death in vivo (3).
My laboratory conducted in vitro studies with trastuzumab–IR700 bound to HER2-expressing NIH3T3 cells (3T3–HER2) and panitumumab–IR700 bound to HER1-expressing A431 cells, which were then irradiated with NIR light using an LED instrument. We used EGFR-negative Balb3T3 cells as controls. In vivo studies involved mice with 3T3–HER2 and A431 xenograft tumors tested one day after intravenous administration of MAb-IR700 followed by transcutaneous NIR light exposure from an LED.
MAb-IR700 demonstrated specific binding to target receptors on the cell membrane, which was followed by gradual internalization into endolysosomal compartments. MAb-IR700–bound, target-specific cell death could be induced within two minutes of exposure to NIR light, which caused cellular swelling, bleb formation, and rupture of vesicles that indicated rapid, necrotic cell death. We observed no phototoxicity in cocultured, receptor-negative Balb3T3 cells after incubation with MAb-IR700, even when the MAb-IR700 was not removed from the medium during light exposure.
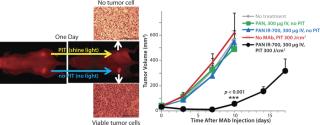
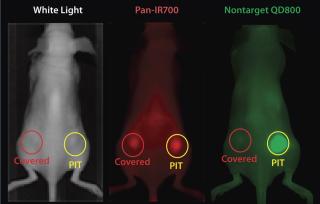
EGFR target-specific MAb-IR700 accumulated in the mouse xenograft tumors, as shown on NIR fluorescence images (Figure 5). We measured >90% tumor shrinkage within three days of the NIR irradiation with no apparent side effects — but only in the EGFR-expressing tumors.
Because IR700 emitted weak but visible fluorescence, we found therapeutic light delivery to be highly directable to target tumors. Furthermore, this allowed for monitoring after therapy as fluorescence decreased with cell killing. The MAb-IR700 PIT was most effective when it was bound to cell membranes, but it showed no phototoxicity when unbound, suggesting a novel mechanism for PIT compared with conventional PDT (3).
PIT using panitumumab-IR700 conjugates targeted to epidermal growth-factor receptor (HER1) followed by a single exposure to NIR light in HER1-expressing tumor models (A431) was effective in reducing tumor burden without significant side effects. Tumor recurrences were common, however. One possible reason for that recurrence would be inhomogeneous intratumoral microdistribution of the panitumumab-IR700 conjugate leading to regionally ineffective PIT. By fractionating the administration of the panitumumab-IR700 conjugate, followed by multiple repeated NIR light exposures to the tumor, we achieved dramatically better tumor control as the right panel in Figure 5 shows (11). We demonstrated (Figure 6) that the effect was related to improved antibody redistribution into the remaining tumor as a result of superenhanced permeability and retention effects (SUPR) induced by the first PIT treatment (12). Most A431 tumors were eradicated with repeated PIT, again without apparent side effects, and the mice survived tumor-free for over 90 days. We monitored therapeutic effects using IR700 fluorescent signal.
Most cancer cells are deformed and killed immediately upon exposure to NIR light, as demonstrated by recent bioluminescence imaging experiments (13). Whereas cytotoxic effects are nearly immediate, though, overall tumor size remains unchanged for one or two days before dead cells are removed by macrophages and the tumor size decreases. Therefore, new imaging methods will be required to monitor early tumor response in the immediate period following PIT treatment.
Although IR700 is a weak fluorophore, it does have the interesting property that its fluorescence life time increases once IR700 is internalized and endocytosed in celluar lysosomes. During NIR light exposure, the lysosomal membrane is also disrupted together with the cell membrane, which releases IR700 into the cytoplasm and shortens its fluorescence lifetime. That shortening of the fluorescence life-time could be a good biomarker of cell killing (14), and fluorescence life-time measurements could be used to measure the effects of PIT on tumors.
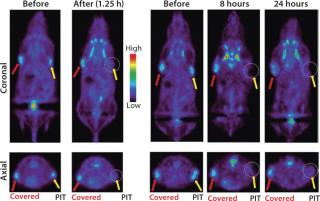
When treated tumors are not visible from the surface of an animal, other imaging methods must be considered for evaluating the immediate effects of PIT. We have investigated the role of positron emission tomography (PET) for evaluating the metabolism of tumors treated with PIT. Uptake of 18F-FDG (SUV) was reduced 85% immediately after NIR light exposure, as Figure 7 shows, and it remained low at all time points as tumors decreased in size (15).
By merging photonics technology with cancer targeting antibodies through immunoconjugation, we successfully developed sensitive and specific imaging methods for cancer detection that are feasible for clinical applications. In addition, with a similar strategy, we established a highly specific cancer therapy approach that can specifically and potently kill cancer cells by pure necrosis induced by NIR light exposure. This unique method of cell killing can enhance the effects of nano-sized or macromolecular drug delivery, and it could elicit an improved host immunity to cancer.
Author Details
Hisataka Kobayashi, MD, PhD, is chief scientist inthe molecular imaging program at the US National Institutes of Health’s National Cancer Institute, Building 10, Room B3B69, MSC1088, Bethesda, MD 20892-1088; 1-301-435-4086, fax 1-301-402-3191;
REFERENCES
4-609.