
Fotoproduktion bei Sartorius, Göttingen vom 04.12.-10.12.11
Increased commercial use of single-use systems (SUS) for large-scale biopharmaceutical production creates the need for consensus on industry best practices and standards for materials in SUS components. End users and suppliers are beginning to develop a shared vision of industry needs in such areas (1, 2). For example, highly visible efforts to harmonize extractables testing include contributions from groups such as the BioPhorum Operations Group (BPOG), Bio-Process Systems Alliance (BPSA), Parenteral Drug Association (PDA), ASTM, and ISPE.
In addition to extractables testing, similar collaborative efforts focus on particulates, integrity testing, interchangeability of materials/components, and supplier qualification requirements. We build on this vein of current industry work, discussions, and recent publications by proposing development of a new standard assay for assessing the suitability of specific plastics in cell-growth applications for SUS (3). This type of testing, similar to the incorporation of other consensus standards testing in the SUS qualification process, would reduce the in-use risk of SUS technology and assist in new product development and change management.
Demonstrating Suitability
Suppliers of single-use bioreactors (SUB) and media/buffer containers used in good manufacturing process (GMP) applications have historically applied USP <87> and USP <88> testing to demonstrate suitability for the application as part of their product documentation. However, those standards were originally developed for medical device applications that are different from modern serum- and protein-free cell culture applications. Recent industry reports demonstrate that current compendial biocompatibility testing did not always detect cell growth issues (4–6). Discovery of the cytotoxic leachate bis(2,4-di-tert-butylphenyl)phosphate (bDtBPP) (6) raised industry awareness of the need for new evaluation techniques and testing to determine potential impacts of plastics used in SUS on cell culture processes.
The knowledge of factors interfering with cell growth and product formation is still limited. Observed issues can be highly cell-line dependent (e.g., sensitivity to bDtBPP, cholesterol dependency, hormone-like effects of slip agents, and so on). The recent awareness of the impact of leachates in cell culture operations highlights the need for suppliers and end users to share a technical understanding and transparency on the procedures for SUS raw material and process changes. A well-designed change-control process will reduce the risk of changes affecting cell growth behavior and/or final product quality. As the industry works to develop best practices for supply-chain change-control processes (7), the development of a standardized assay will enable evaluation and quantification of changes to cell growth.
A Standardized Cell-Growth Assay (CGA)
In this context, a standardized CGA would provide an industry-wide, aligned, and reliable approach to testing the suitability of single-use bioreactors and media containers for specific applications. Furthermore, a standardized CGA would be useful to SUS suppliers for development and qualification of new materials and components for demanding SUS applications. The assay would be an essential enabler of a quality-by-design (QbD) approach to selecting materials, specifications, and process controls based on chemical and/or physical analytical methods. Used as a tool in the understanding of process effects on product performance, a CGA could then be extended to support
- the release of critical raw material/component batches if process understanding and/or controls are insufficient to demonstrate suitability
- qualification of changes to critical raw materials and/or processing parameters, and
- periodic retesting of critical raw material batches as additional controls.
From an end-user perspective, standard CGA test results in an equipment validation package would enhance assurance that the SUS containers meet their basic process requirements.
The CGA should involve incubation of cell growth medium in suitably scaled test articles under defined conditions of temperature, time, and ratio of container surface area to media volume. The treated medium would then be transferred to standard containers (plates or shake flasks, for example), inoculated with a specified density of cells, and incubated, with cell growth observed after a specified period of time (and, potentially, number of passages). Growth parameters should be normalized to controls that are run using media incubated under identical conditions but in containers known to be inert. The data outputs would thus provide values for (normalized) viable cell density as well as viability for each test article.
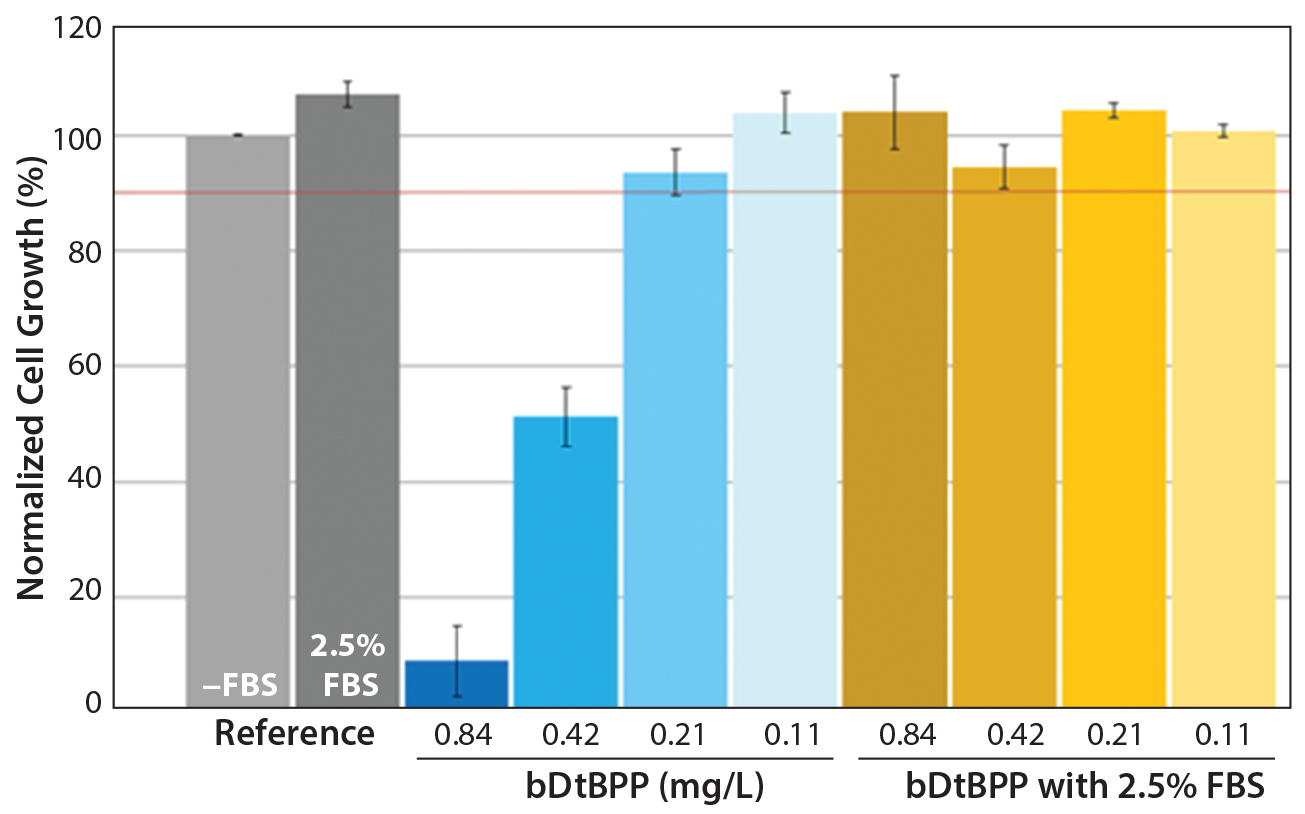
Figure 1: Cell growth results using a Chinese hamster ovary (CHO) cell line and experimental system (8) show that fetal bovine serum (FBS) can mask the cytotoxic effects of bDtBPP. Spiked bDtBPP concentrations in the range of 0.11–0.84 mg/L are compared with unspiked (reference) conditions.
Cell-Line Identification: Of course, the element most crucial to the CGA is identification of an appropriate cell line. The first and most obvious requirement is that the cell line be freely available for purchase and not subject to intellectual property or other restrictions for this use. Further, the growth medium should be chemically defined, with published composition. Addition of serum or hydrolysates to the medium would potentially mask the effects of cytotoxic leachables or otherwise decrease the sensitivity of the assay. For example, working with a Chinese hamster ovary (CHO) cell line previously used at one of our companies (SSB) (8), we have seen that even concentrations of serum below 2.5% can negate the cytotoxic effect of bDtBPP (Figure 1).
The Influence of Process Conditions: Further investigations at SSB are ongoing to better understand the influence of process conditions on this (proprietary) cell line, and similar efforts are undoubtedly occurring at other companies. Given the amount of time required to qualify a cell line and corresponding assays, it would be beneficial to establish an industry-accepted (and nonproprietary) cell line to reduce the amount of double work.
Because the scope of the CGA does not include evaluation of product titer or product quality attributes, it is not required that the chosen cell line be a genetically engineered production cell line. In our opinion, CHO cells are the preferred cell type, given the widespread use of CHO in biopharmaceutical production and the documented sensitivity of several CHO cell lines to SUS materials. Because bDtBPP cytotoxicity is a known risk in SUS, the chosen cell line should be sufficiently sensitive to that compound to serve as an early warning system.
Currently, scientists at our companies are assessing the suitability of the CHO XM111-10 cell line: Recent reports by DECHEMA (Germany’s Society for Chemical Engineering and Biotechnology) show different rates of cell growth in separate types of disposable bags (5). Results from that report are qualitatively consistent with those obtained from our respective in-house cell growth testing. The sensitivity of CHO XM111-10 to bDtBPP and several other known cytotoxic compounds is being explicitly assessed. Among the latter are compounds known to interfere with cellular DNA synthesis and energy metabolism. Admittedly, there is no guarantee that heretofore unknown problematic leachables will be effectively detected. Nonetheless, a CGA using a generally sensitive CHO cell line represents a significant improvement from the current testing regimen.
Selecting Other Assay Parameters
Whatever specific cell line is selected, several other assay details also must be established. Assay parameters such as incubation time, ratio of specimen surface area to medium volume, and inoculation seed density will need to be fixed. They should, however, be chosen such that the assay clearly demonstrates that materials already known to pose process risks actually do so.
Water and Media: Because water and media can have different extractive power toward leachable compounds (9), medium should be incubated directly in contact with a test article rather than made from water incubated with it. For testing finished containers, this would be best accomplished with the medium in the container itself. For individual material screening, the test material should be placed with medium in an inert (e.g., glass) container. Directly incubating media also retains the possibility of detecting potential problems due to selective adsorption or absorption of key media components by the test article (10).
Test articles should be assayed promptly after sterilization because improvement in a bag’s performance in an in-house CGA with increasing time after gamma irradiation has been reported (8).
Specimen preparation must be well defined. Formulations of allegedly identical base materials (e.g., polyethylene) can vary with respect to added processing aids or adventitious contaminants, depending on whether the final component is being converted by (e.g.) injection molding, machining, extrusion, or some other process.
The geometry of the specimen needs to be accounted for (e.g., whether tube-like material or flat sheets). With assay parameters set, robustness studies also will need to be conducted to assess the repeatability of results and the ability of different laboratories with different equipment (within reason) to achieve comparable results.
Practical Considerations: Although the assay parameters must be fixed and limits set to decide whether the test outcome is valid, it is not advisable for the official standard to specify rigid accept–reject criteria for the use of materials or assemblies being tested. Suppliers and end-users should be able to interpret results and establish action thresholds individually. As a practical matter, it is unnecessary that the standard CGA be used for lot release of final SUS product.
Finally, it should be stressed that this assay would be relevant only to upstream bioprocesses. It would not be suitable for testing downstream processes that present other patient safety concerns. For this reason, we propose that the eventual CGA standard be developed and adopted through a consensus standards organization such as ASTM rather than through a pharmacopoeia.
The Importance of a Life-Cycle Approach
In addition to qualification of raw materials, a CGA can serve as a reference system for routine monitoring of critical components/system throughout their life cycles, potentially accelerating the root-cause analysis for both suppliers and manufacturers if process performance is unexpectedly altered by an unknown factor. Given the significant potential utility of a standard CGA to stakeholders — including suppliers and end-users — we advocate robust public discussion on this topic.
References
1 Whitford W, Galliher P. Trends in Setting Single-Use Technology Standards. BioProcess Int. 12(9) 2014: 12–17.
2 Vogel JD, Eustis M. The Single-Use Watering Hole: Where Innovation Needs Harmonization, Collaboration, and Standardization. BioProcess Int. 13(1) 2015: insert.
3 Martin J. Cytotoxicity Risks in SingleUse Media and Bioreactor Containers. BioPharm Int. 27(2) 2014: 16–18.
4 Horvath B, et al. A Generic Growth Test Method for Improving Quality Control of Disposables in Industrial Cell Culture. BioPharm Int. 12(6) 2013: 34–41.
5 Eibl R, et al. Recommendation for Leachable Studies: Standardized Cell Culture Test for the Early Identification of Critical Films for CHO Cell Lines in Chemically Defined Culture Media. DECHEMA Biotechnologie 2014; www.dechema.de/en/posis.html.
6 Hammond M, et al. Identification of a Leachable Compound Detrimental to Cell Growth in Single-Use Bioprocess Containers. PDA J. Pharm. Sci. Technol. 67(2) 2013: 123– 134.
7 Kline S, et al. Change Notifications for Single Use Components: Criteria from an End User Perspective. Pharm. Engr. 34(3) 2014: 1–8.
8 Jurkiewicz E, et al. Verification of a New Biocompatible Single-Use Film Formulation with Optimized Additive Content for Multiple Bioprocess Applications. Biotechnol. Prog. 30(5) 2014: 1171–1176.
9 Hammond M, et al. A Cytotoxic Leachable Compound from Single-Use Bioprocess Equipment That Causes Poor Cell Growth Performance. Biotechnol. Prog. 30(2) 2014: 332–337.
10 Kadarusman J, et al. Growing Cholesterol-Dependent NS0 Myeloma Cell Line in the Wave Bioreactor System: Overcoming Cholesterol-Polymer Interaction By Using Pretreated Polymer or Inert Fluorinated Ethylene Propylene. Biotechnol. Prog. 21(4) 2005: 1341–1346.
Corresponding author Alexander Tappe is director of laboratory cell culture in R&D at Sartorius Stedim Biotech GmbH (Goettingen, Germany); Alexander.Tappe@ sartorius-stedim.com. Jonathan Cutting is director, R&D, North America, at Sartorius Stedim North America, Inc. Matthew Hammond is a senior scientist in core technologies at Amgen Inc. (Thousand Oaks CA, USA); Heather Nunn is a scientist in drug-substance technologies at Amgen Inc. (Cambridge MA, USA), and Sally Kline is director of materials science at Amgen Inc. (Cambridge MA, USA).