With the help of rapid advancements in molecular biology and genetic engineering, a rising number of biotherapeutics are being developed and marketed. The quality and safety requirements for this class of active ingredients has steadily increased over the decades since Eli Lilly put forth the first insulin manufactured using genetically modified organisms in 1982. This has led to dynamic developments in protein analysis and proteomics intended to meet a growing demand for new technologies and sophisticated analytical techniques to characterize therapeutic proteins.
Meanwhile this new class of drug substances has posed great challenges to regulatory authorities and the pharmaceutical industry, both of which were previously used to dealing primarily with small-molecule drugs. Proteins are much more complex entities, and as a consequence, a complicated set of rules has developed to guide the approval of biotherapeutics. Discussion about regulatory requirements concerning therapeutic proteins has not yet abated. Each protein poses very individual challenges depending on its structure and mode of action. And although our knowledge of proteins and their function is steadily increasing, it is by no means complete.
To highlight the complexity of these issues, it is useful to review a relatively simple technique, such as the quantification of proteins, as an example method that must be carried out to a standard that enables approval by the regulatory authorities. Quantification of proteins is a basic requirement throughout drug development, from drug discovery to release testing. Providing an exact and reproducible drug dose to a patient is critical for increasing drug safety and ensuring efficacy. But the challenge remains to develop a precise, robust, and reproducible method of quantifying drug dosage. Currently, no single method of quantification can determine the true protein concentration for all proteins in all kinds of buffers to serve as a gold standard.
The lack of a “gold standard” method is attributable to the large variety of structures proteins can have. This variety in turn leads to a large variation in physicochemical properties and microheterogeneity of these products, and it hinders the universal quantification of proteins. The goal for protein quantification in the pharmaceutical industry must therefore be to develop robust and validated methods suitable for each individual protein in its specific matrix. And to reach that goal, the correct choice of quantification method is crucial.

WWW.PROTAGEN.DE
Three main aspects have to be considered when choosing the correct protein quantification assay: where it will be used, which protein will be quantified, and what that protein’s unique properties are.
Where in Drug Development Will the Assay Be Used? During drug discovery, a drug substance is usually available only in low quantities and at low purities, and it is often not well characterized. Here, fast and flexible methods of high-sensitivity quantification are required. Methods such as Lowry, BCA, amino-acid analysis, and simple one-dimensional (1D) gels are commonly used. For host-cell protein detection, commercially available ELISA kits are applied. As a drug candidate moves toward clinical testing, the active ingredient becomes available in increasingly larger quantities and at higher levels of purity. At later stages, the quantification of impurities increases in importance. For impurity detection and quantification, 1D and 2D gels with silver or Coomassie staining, HPLC with UV detection, and ELISA kits are commonly used.
A drug product is most often administered to patients as part of a formulation. So at this point the main challenge is often to quantify the drug substance when it is mixed with an excess of matrix components. Here, very robust and reliable assays such as amido-black can be used. Exact quantification of the drug substance is often required at this stage. Amino acid analysis offers very reproducible results, although care must be taken to include the buffer as a negative control and account for possible interference. And for release testing of clinical trials materials, a major consideration is whether an assay is described in the European, US, and/or other pharmacopoeia (1). If so, the assay will be more easily accepted by the regulatory authorities.
What Needs to Be Quantified? The goal of a protein quantification assay should be defined as early as possible. Whether the drug substance alone or the total protein content of the sample needs to be quantified will determine which assays can be considered. If impurities are to be quantified, it is necessary to ask whether just the major components need to be analyzed separately or whether the sum of all the impurities should be simply determined. Colorimetric assays generally quantify all proteins in a mixture, giving its total protein content. Assay methods specific for the drug substance include ELISA, Western blots, and mass spectrometry (AQUA). It may, however, be advisable to quantify each impurity separately. If so, a 1D gel with Coomassie staining will resolve each individual impurity for separate quantification of each substance. If the drug substance alone is to be quantified, then to what extent should possible isoforms or degradation products be quantified as well or discriminated against? This is especially difficult to assess for ELISA assays, in which specificity of antibody binding is not readily defined. New tools such as protein biochips can help determine the specificity of antibodies used. For quantification by mass spectrometry, specificity can be determined to a certain extent by a thorough bioinformatic selection of suitable peptides for analysis.
What Are the Unique Properties of the Protein of Interest? Last but not least, the individual properties of the protein to be quantified will influence the choice of assay. Besides important matters such as available amounts and purities, protein parameters such as solubility, molecular weight, isoelectric point, amino acid composition, stability, and homogeneity must be considered. Buffer components and other excipients should be taken into account as well.
For example, low protein solubility in aqueous solutions can make the use of detergents or chaotrope reagents necessary, and those can interfere with many protein quantification assays. A robust assay (e.g., amido-black) may improve the reliability of quantification. Labile proteins and proteases can lead to substantial degradation during sample preparation and thus makes reliable quantification impossible. Deactivating proteases, for example by denaturation in urea, may be an option but only if the assay can tolerate it.
Due to all those factors, choosing the best protein quantification assay is not straightforward and requires experience in the field. Table 1 summarizes the most important parameters for several of the most widely used protein quantification assays.
Table 1A: Comparing the main characteristics of protein quantification assays, beginning with tolerance for reducing and chelating agents, detergents, salts, and saccharides
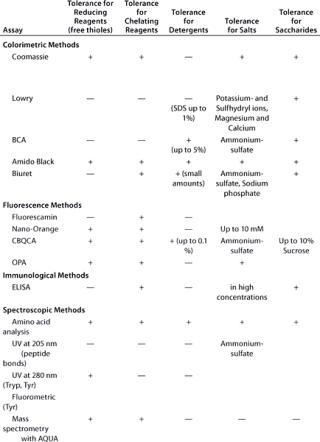
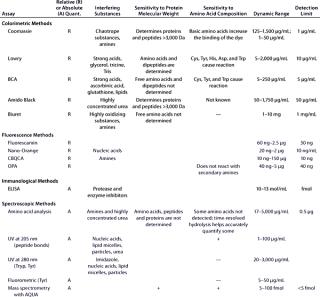
Table 1B: Comparing the main characteristics of protein quantification assays, including relative (R) or absolute (A) quantification; interfering substances; sensitivity to protein molecular weight and amino acid composition; dynamic range; and detection limits
Once you’ve chosen and established a protein quantification assay for your specific application, the next step is to validate that assay for use (e.g., in investigational new drug submissions or lot-release testing). One efficient approach is to use a risk-based validation strategy. Risk analysis techniques such as FMEA (failure mode and effect analysis) have proven useful in this regard.
The first step of this type of validation effort is for a team of experienced researchers and technicians to conduct a risk assessment. All steps in the assay, from sample generation to data analysis, are analyzed using a systematic approach and then categorized objectively. The goal of this detailed analysis is to assess the influence that each step will have on the final result of an analysis and to rank those steps according to the risk of causing the result to be wrongly determined.
The FMEA result is a list of parameters that must be evaluated to obtain a robust assay. Therefore, robustness analysis is the next logical step toward validation. Those parameters ranked as critical to determining the result of an analysis are systematically studied. If small changes to one of them have a large influence on the analysis result, then that parameter will subsequently need to be monitored very closely. Measures will be taken to ensure that the assay will deliver reliable and correct results during, for example, release testing. Such measures could include adding comments to a standard operating procedure (SOP) or test procedure (e.g., “Tolerances must be tightened for critical volumes.”) or involve the qualification of system components (e.g., HPLC columns). If measures that can be implemented do not address all the critical steps of a protein quantification assay, that assay cannot be validated.
Validation can begin only if a sufficiently robust assay can be established. For methods already described in appropriate pharmacopoeia, the robustness test can be cut short because the validity of those assays is generally assumed to be sufficient (2). In such cases, the only verification required is to show that a user can conduct an assay as described and that the assay is indeed suited for its application. The pharmacopoeial methods have the additional advantage that regulatory authorities are familiar with them already, which in many cases will simplify approval.
The bases for the method verification/validation that follows are risk analyses and instrument/software qualification. For protein quantification, the choice of an appropriate reference standard is crucial because in most cases, a relative quantification is carried out against that standard. To establish a valid assay, the reference standard has to meet at least these criteria:
-
Both sample and reference standard are in good agreement with respect to protein composition, modifications, matrix substances, and concentration.
-
If levels of protein contaminants or impurities are to be quantified alongside a drug substance, the reference standard must represent the mixture of proteins in a sample. In some cases, it may be easier to quantify each impurity separately and thus have a number of different reference standards, one for each impurity.
-
If contaminations or impurities are to be quantified, a reference standard is representative of the mixture of proteins in the sample. It may be better to quantify each impurity separately.
-
Concentration of the reference standard is determined as well as possible. This can be done by using a primary reference standard (e.g., from the National Institute of Standards and Technology, NIST, or the European Directorate for the Quality of Medicines, EDQM) to calibrate a secondary reference standard or by using an alternative quantification method. The latter may be inaccurate, however. Different quantification methods produce different results because of specific systematic errors associated with each type of assay (e.g., colorimetric assays stain peptides only above a certain size, and amino acid analysis also quantifies peptides and free amino acids).
-
The standard must be stable for a longer period than the sample.
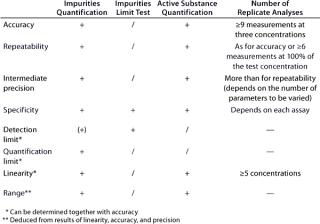
According to an ICH guideline, the following parameters have to be validated for a quantitative assay: accuracy, precision, repeatability, intermediate precision, specificity, detection limit, quantification limit, linearity, and range (3). If only a limit test is conducted (for assessing the presence of impurities), then the validation of specificity and detection limit will suffice. Table 2 provides an overview of the regulatory requirements, and the “Definitions” box defines the terms listed above.
DEFINITIONS OF ICH VALIDATION PARAMETERS
Accuracy expresses how closely the value accepted either as a conventional true value or an accepted reference value and the value found agree.
Precision expresses the closeness of agreement (degree of scatter) between measurements obtained from multiple sampling of the same homogeneous sample under prescribed conditions.
Repeatability expresses precision under the same operating conditions over a short interval (also termed intra-assay precision).
Intermediate precision expresses within-laboratory variation (e.g., among different days, analysts, equipment).
Specificity is the ability to assess unequivocally an analyte in the presence of components that may be expected to be present.
Detection limit is the lowest amount of analyte in a sample that can be detected but not necessarily quantified
Quantification limit is the lowest amount of analyte that can be quantitatively determined in a sample with suitable precision and accuracy.
Linearity describes the ability to obtain test results directly proportional to the concentration of analyte in a sample.
Range is the interval between upper and lower quantification limits.
Table 2: Validation parameters according to ICH (“ + ” = paramet
er must be validated; “/” = no need for validation; “(+)” = must be validated, if applicable)
At Protagen AG, we validated an assay for protein quantification (according to the ICH guideline mentioned above) using a tumor lysate as a model matrix and bovine serum albumin (BSA) as a model protein. The goal was to develop a robust assay that could quantify the total protein content of samples in a wide variety of buffers. The assay was intended to be used in earlier stages of drug development (in which quantification of samples is carried out in different buffer conditions for optimization) and for release testing, in which formulation excipients can interfere. The assay needed to quantify all proteins in a sample because a drug substance can be a complex mixture of proteins.
The assay we chose was a modified protein-amidoblack-complex precipitation using triplicate determinations based on a published method (4, 5). Our modifications mainly revolved around the introduction of a 96-well plate fitted with a membrane bottom, which allowed us to reproducibly isolate the protein-dye precipitate. Protein quantification was carried out relative to a BSA standard curve.
To evaluate critical steps, we performed an FMEA. One critical step we found was using commercially available BSA without checking the actual concentration of each lot. The measure we took to overcome this problem was to use a NIST standard to check protein concentrations for the calibration curve. Another critical point was found around the use of the multiwell plates for the assay read-out. Each lot of plates had to be checked for homogeneous read-out in all well positions. Also, the calibration standard and analyte were dissolved in the same buffer for this assay. SOPs describing all these relevant issues were written and are being used by Protagen technicians to carry out tests under GMP conditions on a range of therapeutic proteins.
With those and other outcomes of the FMEA addressed, the robustness of this assay could be considerably increased. Therefore, the validation characteristics of linearity, accuracy, precision (both repeatability and intermediate precision), detection limit, quantification limit, and range could be determined.
Robustness and Specificity: Here, our main concern was to measure only the protein content of a sample and prevent the influence of buffer components on quantification results. For proof of specificity, we compared the results obtained from a placebo (dilution buffer), a spiked placebo, and a sample solution. In most cases, buffer composition showed no influence on the quantification results for the buffers tested (Figure 1). In each case, concentration of the spiked protein was correctly determined.
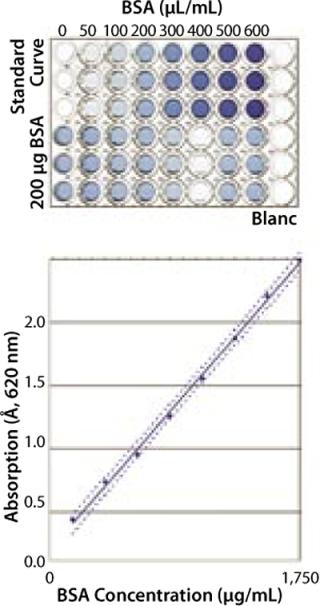
Linearity: We measured absorption of eight BSA samples over a range from 100 µg/mL to 1,750 µg/mL to determine linearity of the assay. The resulting optical density (OD) was directly proportional to protein concentration in this range (data not shown).
Accuracy was shown by spiking tumor lysates of known concentration with defined amounts of BSA and subsequently performing a recovery calculation. The acceptance criterion of 100 ± 5% recovery was well met.
Precision: The coefficient of variation (CV) was shown to be less than 5% for hexaplicate analyses using 500 µg/mL Jurkat cell extractions as sample. This shows the excellent precision of the assay.
Detection and Quantification Limits (DL, QL) were calculated from the standard deviation of a hexaplicate blank (dilution buffer) measurement as well as from the linear dilution curve. The QL was determined to be 40–100 µg/mL.
Validation of the assay has led to a specific plate layout that we now use for all protein quantification analyses. It includes the standard curve in triplicate, the samples in triplicate, and negative controls. One major criterion for accepting the results of these assays is the quality of the reference curve, which is measured by a correlation factor and the slope of the linear fit. Data analysis is carried out using a validated Microsoft Excel spreadsheet.
So this protein quantification method as performed is linear, accurate, and precise, at least within a range from 100 µg/mL to 1,750 µg/mL BSA. This gives a robust and validated method for protein quantification. Our validation shows the main advantages of the method compared with other assays such as Bradford or Lowry, including a large linear range and a very good tolerance for differing buffer compositions.

WWW.PROTAGEN.DE
Developing a validated protein quantification assay for therapeutic proteins and antibodies is a major challenge. When choosing the correct assay, consider its aim (e.g., therapeutic protein or impurities), the nature of the protein of interest (e.g., stability or homogeneity), the matrix substances (e.g., detergents or stabilizing agents), and regulatory requirements (e.g., specificity and detection limit). In most cases, multiple assays have to be used, and each of them must be optimized and validated for one specific part of the testing.
Protein quantification is one of the seemingly easier tasks involved in characterizing a therapeutic protein. But even such a fairly simple assay can be complex when it comes to robustness and validation. Service providers with extensive experience in protein analysis under GMP conditions can help researchers carry out appropriate assays and help drug developers progress their programs.