Most products in discovery by pharmaceutical companies today are biopharmaceuticals. Made by living organisms, these are typically large–molecular-weight products that rely on their secondary and tertiary structure for therapeutic effectiveness. Synthetic small molecules and biopharmaceuticals both require analytical verification for release, but only biopharmaceuticals require functional potency assays for investigational new drug (IND), biological license application (BLA), and new drug application (NDA) submissions. Those activities often require elaborate transfers of diverse, biological, product specific assays that carry greater chances of error. Shortcomings are common in bioassay transfer documentation, planning, and setting timelines, which cause costly processing delays.
Barriers to Success: A common negative attitude is that analytical assays are more important than bioassays and given higher timeline and financial priorities. Another is that a binding assay will do for phase 1 release. Although in certain instances a binding assay may be accepted for phase 1, more often than not the FDA expects a true functional assay as part of the release testing and stability panel for first-in-man studies. Associated product specifications may be simply “Report results,” but the method should be rugged enough to pass scientific and regulatory scrutiny. Another problem occurs when research staff, unfamiliar with the rigors and expectations of the regulated environment, deem a method ready for transfer without thorough validation/optimization and/or detailed documentation. Yet a fourth hurdle may occur when small-molecule project managers set unrealistic timelines that are more appropriate to predictable analytical assays than to complex cellular assays.

Many types of method transfer exist: e.g., from an company’s research laboratory to its GMP laboratory and from one GMP site to another. However, most bioassay transfers occur when companies with non-GMP laboratories outsource their research assays to regulated contract research organizations (CROs). For phase 1 IND candidates, it is most common for scientists to transfer unvalidated bioassays using a method acquisition process and then follow that with optimization and GMP qualification. For the purpose of clarity, here I use the term qualification for phase 1–2 validations. The differences between those and phase 3 validations are well covered elsewhere (1,2).
To initiate all transfers (whether early or late), a transferring organization must prepare comprehensive and accurate documentation detailing its bioassay method. Effective management of this step is critical; regulatory inexperience in biopharmaceuticals and underestimating the importance of a well-documented transfer cause significant problems. Lack of attention to this step causes roadblocks in development and can delay the fast track of a company’s commercial objectives sometimes for weeks or months. Well-documented methods will save time and money and lead to greater success in bioassay production.
Preparing for an Effective TransferPartnering with the Right CRO: To help ensure effective transfers, select an appropriate, well-qualified CRO that is experienced with your type of biologic product. In your evaluation, include the following questions.
-
How experienced is the CRO in developing, validating, and processing bioassays? Bioassays are very diverse in their mechanisms and endpoints, so the organization should have at least five years of experience.
-
Does the CRO have state-of-the-art technology and a high-quality systems infrastructure? The sensitivity and endpoints of its detection instruments should be method appropriate according to current industry and regulatory expectations. Also, at least four data analysis software systems should be available to properly evaluate curve similarity and sample potency. Compliance Wire, Trackwise, Documentum, and other electronic systems help assure compliance in all aspects of a project.
-
How many INDs, BLAs, and NDAs has the company supported? If a company has been involved in at least 10 INDs and 2 BLAs/NDAs, it should have enough organizational experience to maturely answer regulatory questions and support audits.
-
Is the bioassay group a separate organization under a director or merely an add-on to the analytical group? If a method requires development or troubleshooting, then a CRO with seasoned senior management should be a major consideration. Other factors (e.g., proximity and price) may be built into decision making if a company is transferring a robust and fully validated bioassay for routine sample analysis.
-
What is the ratio of doctorate level to nondoctorate scientists in that group? At minimum it should be 30/70 while a method is in development; 10/90 is sufficient once the method proceeds to sample analysis and routine maintenance.
-
What is the facility’s FDA Form 483 audit history? Clearly, no 483 history would be preferred. But any 483s should have been only occasional (no more than two or three in a 10-year period) and minor as well as quickly corrected.
For an effective CRO partnership, outsourcing professionals should establish clear project parameters, set realistic timelines and expectations for deliverables, develop and manage a productive plan for communications between finance and operations, and negotiate and execute the best-value financial arrangement.
Is the Method Transferred or Acquired? A common problem in the method transfer process occurs when research scientists request a transfer, but in reality a “true” method is not available. A true, validated (or at least qualified) method needs to meet specific minimum system suitability requirements — such as accuracy, precision, minimum response, and parallelism — that are lacking in many non-GMP methods. Acquired methods typically have not undergone a thorough ruggedness/robustness validation in the GMP environment and may not be readily reproducible when executed in a different environment. Normally the acquisition phase will be followed by an optimization phase, and the method will need to be validated or at least qualified again before it can be used for GMP work. The process allows an acquiring lab to evaluate gaps and include important specification parameters for system suitability and a qualification protocol.
True transfer activities should be limited to methods that have been validated for robustness or at the very least qualified under GMP conditions. However, a non-GMP familiarization phase is recommended for all cell-based assays so that scientists can fully understand a cell line before starting on the related GMP transfer protocol. The minimum requirements for a true GMP cell-based assay transfer are a master cell bank (MCB), a working cell bank (WCB), a well-written method, a transfer protocol, and fully qualified biological reagents.
The familiarization phase should consist of at minimum two assays executed by two analysts on two different days using a freshly thawed vial from the working cell bank. If more than one assay fails, familiarization continues until at least two consecutive assays pass per analyst. For some cases in which the equipment or software of two organizations is significantly different, some minor optimization followed by a small cross-validation may be required. Once all analysts are trained, a minimum of one assay executed by two of them on two different days will be needed for GMP transfer.
Acquired (non-GMP) assays that have not undergone validation or qualification use the same familiarization procedure as a GMP assay. However, if more than 25% of these assays fail, a project has to proceed to an optimization phase. Once an assay is considered rugged, several analysts must execute it several times to confirm its specifications for the qualification or validation protocol. For a phase 1–2 qualification, two analysts are considered sufficient; three or more should be involved in a phase 3 validation.
Setting a Realistic Timetable: When planning your timeline, first determine how many assays you can run per week. Vast differences in assay procedures greatly affect these timelines. For example, a potency bioassay for a cytotoxic agent can take only two days: one to incubate cells with the drug and one with a viability stain to measure cell kill. However, a cytokine-induction bioassay can take up to a full week or more: one or two days to differentiate cells, one day to treat with the drug, and an additional day to assay supernatant for the specific cytokine. This kind of information needs to be provided by the transferring organization to accurately predict a timeline according to Figure 1.
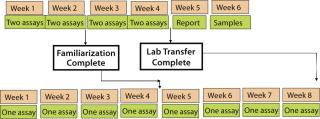
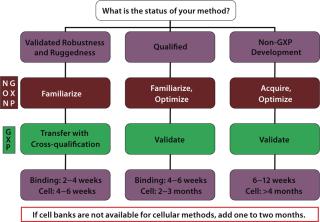
Here’s the rule of thumb: If you run two assays per week, the minimum transfer will take four to six weeks. If you run one assay per week, the minimum transfer will take eight to ten weeks. If GMP-qualified MCB and WCB are not available, add one month to your timeline. Additional time may be needed if preassay conditions have not been set or if a well-written method with templates and worksheets has not been developed. There are also many additional permutations to the transfer process depending on development phase and state of the method. Figure 2 demonstrates how the differences can affect overall time. Setting correct timelines involves plugging the correct information into Figures 1 and 2 for a particular method. (Binding assays are included as a point of reference.)
The method document is the major source of delays in method transfer. Rule number one is to send the right method. That may sound obvious, but products (and their subsequent bioassay methods) may change companies several times, so their histories can become cloudy. Non-GMP labs normally do not have (or need) change control, so many of their transfers to GMP labs are accompanied by methods that are not current or are missing critical pieces of information. Make sure your document lists reagents, current suppliers, and clear expiration dates; describes stability conditions; and is specific about cell maintenance. In my experience, simple overlooked details cause longer delays than obvious errors that can be easily corrected.
To further prevent transfer problems, a scientist in the transferring laboratory who is unfamiliar with the method should first execute the experiment without supervision while relying solely on the method document. Afterward, clarifications or changes are incorporated into the method as needed. A revised document is then sent to the acquiring laboratory for review and initiation of cell maintenance. The final step involves a visit from a scientist from the transferring organization to execute the first assays in parallel with the scientists who will be running the assay in the new organization.
Here are two examples in which improved method documentation before transfer and following best practice recommendations could have significantly reduced the timeline.
Acquisition of an R&D Method for Preclinical Formulation: A biotech company affiliated with an academic institution submitted a research bioassay for a lytic enzyme with antibacterial applications to its CRO. The method was a half-page paragraph consisting of the following steps:
-
Grow the bacterial culture overnight from a colony on agar.
-
Dilute the culture in the morning, grow for three hours, centrifuge, and resuspend in assay media.
-
Add lytic peptide.
-
Measure turbidity change on OD650 every 15 seconds over 15 minutes.
Experiments at the CRO demonstrated that the assay always worked very well, but the bacteria’s sensitivity to the enzyme varied significantly from week to week. This was acceptable for an R&D laboratory, but a fatal flaw for a stability-indicating assay. CRO scientists prepared a GMP cell bank, standardized the original culture’s density using OD readings, and wrote a true GMP method describing every detail of the experiment. Even after those changes, the originating lab could not reproduce consistent CRO data for its own in-house research studies. An investigation demonstrated that the temperature in the research lab was not controlled (a 25 °C chamber is key to most bioassays because room temperature varies), and the reagents were expired (a scenario that is not allowed in the GMP world).
Transfer of a Well-Documented Method with Missing Information: A biopharmaceutical company submitted a very well-written method describing a mammalian-cell–based growth assay using suspension cells. The assay worked well at the CRO except that a “hook effect” distorted the final data analysis, but was not observed in assays executed in the transferring company. After several failed assays, a scientist visited the CRO to evaluate the issue. As soon as the incubator was opened, the problem was identified and solved. The transferring company grew suspensions horizontally, and the CRO grew them vertically. That small detail was enough to set the transfer timeline back four weeks and could have been avoided by adding one word to the method, by the CRO not assuming that all suspensions are grown vertically, and (most important) having the scientist visit the CRO for the first experiment.
Well-Documented Method WritingThese examples show that well-written, accurate, and complete methods will save valuable time regardless of a project’s stage of development. A method document should include the following sections: title, purpose, materials, equipment, reagents, solution preparation and storage, cell maintenance (for cell-based methods), assay procedure, assay acceptance criteria, sample acceptance criteria, and out-of-specification (OOS) procedures. A potency specification should have been defined in the sample analysis protocol depending on the use of the method.
For the best results, first assume that the person performing a bioassay is proficient in the skills required to execute the method but knows nothing about the method itself. Do not abbreviate unless abbreviations are clarified earlier in the document. Finally, keep the document fluid. Instead of referring readers back and forth to other sections, copy and paste those appropriate earlier sections into the later sections that reference them to reduce page-turning and confusion.
Method Title: The title should be project-specific and unambiguous. Instead of referring to a “monoclonal antibody,” state the unique antibody name. The title should also name the specific technology used — “Growth Inhibition of ABC Cells By Alamar Blue” rather than “Growth Inhibition Assay” alone — and state what will be tested, such as a drug substance or drug product.
Purpose: In no more than one paragraph, write a detailed description of the method and its rationale, using references as required and referring to protocols used during development by their work numbers. Call attention to all risky parts of the method. For example, “the assay depends on transfected cells being grown under proper conditions,” “maintenance growth factor lots need to be prequalified,” or “cell checks on thawed cells are required to ensure consistency.”
Materials, Reagents, Instruments: For cell bioassays, name the cell banking protocol number and original cell supplier (if applicable), then specify appropriate cell passages that can be used for the assay. Only the transferring organization can provide the numbers of acceptable passages; failure to correctly supply this information is also a source of delay. Many cell lines have to be in culture for two to three passages after thawing before being optimal for assay execution. Alternatively, some cell lines reach a passage at which they are no longer suitable for executing an assay.
Specify catalog number and supplier of all biological reagents, and be very specific about their type so you can replace a product if the supplier stops manufacturing it. For example, say “protease-free, tissue-culture grade” or “USP grade” reagent. Instruments must be as specifically identified as possible so their compatibility with those of the CRO can be fully evaluated.
Prequalification: Certain reagents, such as fetal calf serum and growth factors, need to be prequalified to ascertain that different batches will support a given assay. Some (e.g., animal complement or secondary polyclonals) may require interbatch changes in the method. Provide clear directions on prequalification and large-batch storage of critical reagents. Also, clearly state appropriate parts of these prequalifications in the purpose, materials, and media preparation or assay procedure sections of the document; do not assume that they will be understood.
Solution Generation and Storage: In the next section, explain receiving and storage conditions. Do not assume everyone keeps media at 4 °C when it arrives from a supplier or that everyone thaws, deactivates, and aliquots fetal calf serum the same way. Define stability times, and give clear directions on how to dissolve stock preparations: e.g., “Centrifuge lyophilized growth factor XYZ for one minute to remove particles from cap. Resuspend in 0.9% saline to a final of 1 mg/mL, aliquot 50 L per microcentrifuge tube and store at −70 °C for up to one year. Use immediately upon thawing, and throw away unused material.”
Cell Maintenance: Be specific in required steps. Describe a cell-splitting procedure with details such as centrifuge speed, time, and aliquots for cell count. Give details of a resuspension procedure, such as “Aspirate, tap gently to break up pellet, add media, and resuspend with pipette.” Write out the cell-counting procedure (including calculations), and specify the minimum and maximum amounts of cell counts required in the hemocytometer for accurate counting, usually >25 and <100 per square. Set a minimum percent viability limit, usually >95% viable by Trypan Blue exclusion. Specify flask position, vertical or horizontal. Describe the schedule by total volume (cells/mL) per flask:
-
30 mL of 2 × 104/mL per 75-cm2 flask for two-day culture
-
30 mL of 1 × 104/mL per 75-cm2 flask for three-day culture (optional but highly recommended)
-
templates provided to track cell maintenance calculations
-
process charts included to monitor cell growth.
Preassay conditions are the most critical aspect of reproducibility in cell-based assay methods. Specify how cells should be maintained one, two, or three days before the assay. Cells should always be on the same growth phase for a given assay. For example: “Prepare two 150-cm2 flasks on the morning before the assay containing 3 × 104 cells in 50 mL media.”
Formulas: Do not assume everyone applies the same formula. Write out all necessary formulas, even those that are commonly used.
Out-of-Specification (OOS) Results: Some institutions have standard operating procedures (SOPs) for OOS results based on analytical methods that may be used as default if a bioassay is very reproducible. If another OOS strategy is to be used for inconsistent bioassays, then it should be clearly written in the method document.
Additional Recommendations: Include tables or templates for media and reagent preparations, cell maintenance, dilutions of drug, plate format, cell checks, assay/sample acceptance, and the final certificate of analysis. Prepare formal process charts from the familiarization, acquisition, and optimization processes to help set specifications for the formal transfer qualification or validation protocols. Keep these charts going through sample analysis to measure trends that may lead to “the brink of chaos.” The final most important recommendation is to base specifications for all methods and transfer protocols on solid, historical data to statistically demonstrate that an assay is accurate and reproducible.
When You Do Outsource…With all the pressures on biopharmaceutical companies today, the more efficient and transparent their bioassay method transfer processes, the faster their track to regulatory submission. Setting realistic timetables, creating thorough documentation to transfer assays between organizations, and working with highly qualified and experienced CROs are keys to streamlining the production of biologics and accelerating their time to market.