Using contract manufacturing organizations (CMOs) to augment your supply chain is not a new phenomenon in the pharmaceutical industry. One of my first projects in industry involved developing a process for a recombinant protein while manufacturing materials for clinical trials. My team recognized that the company did not have the money to build a plant for manufacturing an unproven product, and it was not bullish to the risk of investing, so we turned to a contract manufacturer in Austria. That was in 1984 — fully three decades ago. Even then, we realized that although the CMO performed the mechanics of making our commercial product, our company still had the responsibility to assure that the work was done right. And we set up systems for necessary oversight.

WWW.PHOTOS.COM
Decades later, I found it surprising when I was working for a small company and confronted by the chief financial officer (CFO). The essence of his argument was that using a CMO for its soon-to-be-launched blockbuster makes it unnecessary for the company to employ its own quality people. To put it in his words: “We’ve outsourced the activity. It’s the CMO’s responsibility.” My response to him: “Whose name is on the label, and who holds the license? Therefore it’s our job to assure that it’s done right. Oh by the way, here are my requisitions for the needed head-count.” I got the required staff, but only after arguing about it. But I had to be persistent. That was 1998.
Even today, I consult with companies and find that same old attitude — not always, but all too often. The requirements for quality agreements have been around for nearly two decades in Europe. In the United States, the Food and Drug Administration (FDA) has always held sponsor companies accountable without formally suggesting a way for them to fulfill that requirement — at least not in writing. So I was pleasantly surprised by the May 2013 issuance of a guidance on quality agreements for public comment (1).

Table 1: Sections of a quality agreement
Whenever a new guidance is issued, I peruse it to see whether there might be anything new that I might have missed in my own operations. Mostly I find that the processes I have developed to meet my business needs are more comprehensive than regulatory guidances. After all, I develop processes for my clients — to help me do my business — and not for regulators. I found that the new FDA guidance contains no surprises at all. It includes recommended actions and activities, and they are clearly written. It also contains scenarios to help readers interpret those recommendations. Remember that guidances are not laws. You do not have to follow them — but you will get less scrutiny from the agency if you do.
The new guidance clearly states that a license holder (the drug sponsor) is ultimately responsible for what happens to outsourced product. But this situation is not at all one-sided. A CMO is responsible for operating under current
good manufacturing practice (CGMP) even if the product sponsor is unclear about what that means. The “Practical Examples” box lists some quoted examples from the FDA guidance. The CMO team cannot simply point a finger and say, “The sponsor did not want me to do that.” They must operate as though the product were their own — no matter whether a CMO is making or testing that product.
The agency also indicates that free-standing documents should have sufficient detail for reviewer use. That is, “We are not telling you all that needs to be there.” The guidance does list some elements that should be included: e.g., effective dates and durations, dispute resolution, and responsibilities (including communication and contacts and change control). But that doesn’t mean that change control is the only element of a quality management system (QMS) that needs to be there.
Practical Examples from the FDA (Paraphrased for Brevity)
A quality agreement does not exempt contracted facilities from CGMP requirements related to the operations they perform, regardless of whether such requirements are specifically discussed in the quality agreement (1):
Case 1: Responsibility for facilities and equipment maintenance and upkeep of a contract facility
Case 2: Responsibility for documenting steps in the manufacturing process
Contract laboratories are contracted facilities subject to CGMP requirements
Case 3: Responsibility for data integrity in laboratory records and test results
Case 4: Responsibility for method validation
The guidance goes on into more detail. In a “key elements” section it describes roles and responsibilities, facility and equipment, materials management, product-specific information, laboratory controls, documentation, and change control. Don’t treat this list as a checklist to be obediently followed, but rather as a thought-provoking starting point. Ask yourself, “What must I define to assure that there will be no misunderstandings?” When you develop your documents, the process must be driven by operations rather than lawyers. You can then use that as a basis for negotiating with the CMO’s quality group. Ultimately, you need a document in a language that everyone involved in your project can understand clearly.
Sections of the Quality Agreement
How does my own standard quality agreement stand up against the new guidance? It is more detailed and comprehensive, covering much more, because it is written for a business rather than for the FDA. I have used documents in table form, made up of bullet lists, and prose with sections (e.g., 1.1.2.a.i). All formats work; it comes down to your preference.
I begin by thinking about what is important to a given project. I get input from procurement, logistics, production, quality control (QC), and other groups. Of course, this process is driven by quality assurance (QA). Sometimes I use my company’s QA group, sometimes the CMO’s quality group — whatever works best in a given situation. Think through operations first, then the QMS. How do you make sure that all activities are performed in compliance with the regulations? Go point by point, and ask who does what and what oversight each activity needs. Be prepared to delegate. And in negotiations, remember to put yourself in another person’s shoes: They often have valid reasons for wanting to do things one way or another.
So what are the suggested elements of a quality agreement? Figure 2 illustrates the major sections that I consider when working with a full-service CMO. Below is more detail on specific areas. You may not need to use all these sections, or you may have other areas that you consider important.
Communication and Reviews: It’s critical to build expectations between sponsor and CMO before you begin. For example, frequent may mean every day to one party and monthly to another one. A complete report to one may be a 20-page document rich with data, whereas to the other, a quick email saying everything would be fine. Define who does what, how frequently, with detailed content descriptions, and the mode for all communications. Keep the number of point-contact people to a minimum. Working at a contract manufacturer at one stage in my career, I found it frustrating to field calls daily from 10 different people all asking the same question. Remember that a “point of contact” means a single point — on both sides. You can expand those contacts if needed and contract them back otherwise. Keep communication controlled.
Batch Numbering and Expiry Dating: I was once stymied when a CMO had a nine-digit numbering system, but my company’s electronic systems (which were not very user friendly) allowed a maximum capacity of only eight digits. Believe it or not, that required an excruciating procedural work-around on our part.
How expiry dating is calculated is not universal. Some companies roll everything backward: For product made on 15th of the month, for calculation purposes it was assumed to have been made on the 1st. As a result, a product with an official three-year expiry actually has between 2 years 11 months and three years. In one case, however, a CMO estimated backward between the 1st and 15th and forward from the 16th to 30th. So the first batch was made and labeled by the CMO in the second half of the month, leading to a product with an out-of-specification expiry date. The devil is in the details.
Raw Materials and Vendors: By operating on a large scale, CMOs undoubtedly have good business dealings with their raw-materials suppliers. Take advantage of that: Delegate to your CMO the entire procurement of raw materials. Make sure their quality is appropriate, but otherwise allow the contractor free rein. Some raw materials may be critical and require fastidious specifications, so keep strict control over them, but most items can and should be handled by the CMO. Imagine a contractor in Europe having to deal with procurement from US suppliers where the client is situated.
Master Batch Records: Yes, it’s your product — but the contractor is making it. Let the outsource team write batch records that you review for technical content and completeness before approving them. It may have to be translated to another language for use. I routinely give my English version to CMOs in countries where other languages are dominant, allowing them to translate and put it into their formats and style. Then I translate it back using an independent translator to ensure that nothing has been lost or modified.
Standard Operating Procedure (SOP) Review and Approval: I evaluate some SOPs after the fact by audit; others I review and approve for tighter control. Keep the latter to a minimum because their document-change procedure will be slowed to an excruciating crawl. Don’t force a CMO to operate under your SOPs unless that is critical. Let that company use its own tried-and-true methods, which you can audit to ensure that they meet your standards.
Specialty Reagents and Standards: For small–molecular-weight drugs, this is less of a problem — but for biotech products, you often have to make your own standards because you’re working with a new molecular entity. It’s almost like manufacturing an additional product. Decide who will do this: Will you transfer the manufacture of standards to your CMO, or will you keep it in-house? If you keep it, make sure you have a system in place to give you plenty of notice when new supplies are needed. Don’t get caught running out. The same goes for specialty reagents: e.g., monoclonal antibodies (MAbs) used in enzyme-linked immunosorbent assays (ELISAs) and binding assays. You don’t want your product to go off the market because somebody never made enough reagents.
Specifications: Again, it’s your product, but the CMO has to be able to make it. So that company has a stake in the product as well. Specifications need to be reviewed by both parties. Before you knuckle under to regulators asking for tighter specifications, consult with your CMO. The outsourced team may be able to present a good argument that could help in your negotiations with the agency. I guarantee that you will end up fighting over who pays for failed batches if you unilaterally change specifications without the CMO’s awareness and agreement.
Out of Specifications (OOS) Results: I always evaluate a CMO’s procedure for responding to OOS results in due diligence and require review (and in most cases, approval) of all changes made to it. You don’t want to get caught with an indefensible procedure.
Deviations and Investigations: I usually want to be informed of all major or critical deviations within 24 hours of their occurrance. I require seeing the investigation summary at least before dispositioning the lot. But I do not need to see every little minor deviation.
Define what is major and critical, and over the first few months project teams on both sides should work closely together to fine-tune those early listings and expectations. I routinely audit at month’s end a list of minor deviations to help fine-tune expectations on both sides. Figure 3 illustrates a scenario for cooperative review and approval.
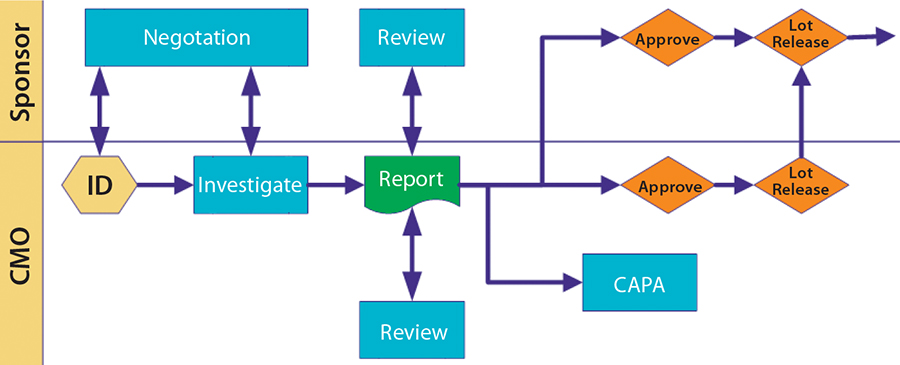
Figure 1: Deviations and investigations
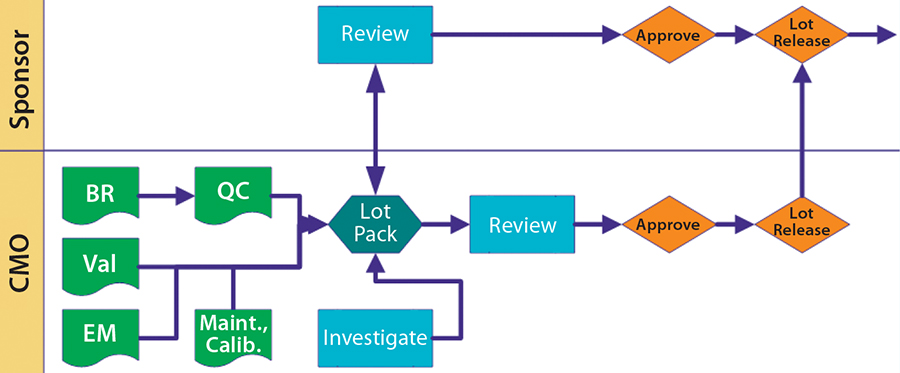
Figure 2: Lot disposition BR = batch record; Val = validate; QC = quality control; Maint. = maintenance; Calib. = calibration
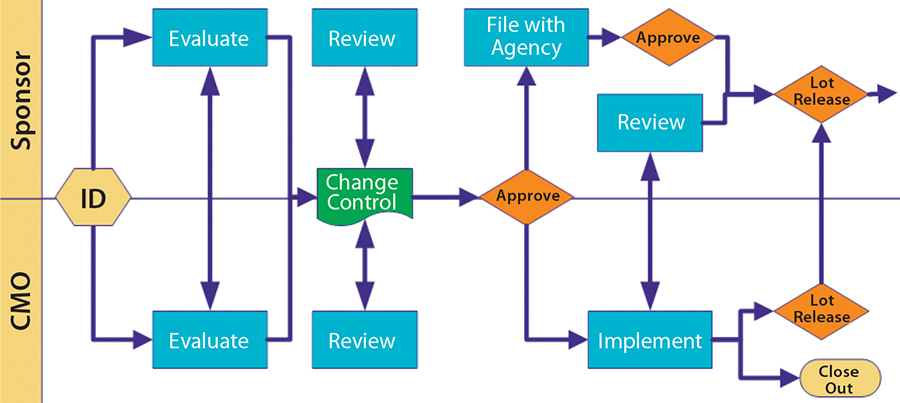
Figure 3: Change control
Batch Disposition: We come back to the product sponsor. Manufacturing disposition is up to the CMO, but the sponsor releases product to clinic or market. A tight and cooperative relationship prevents a “We think it’s okay, but you don’t” situation, at least early on or when a gray area is encountered. Communication is key to keeping such situations from looking messy in documentation.
Figure 2 illustrates possible interactions and relationships. Nothing is written in stone. Just because you begin with tight control doesn’t mean you may not slacken that off somewhat as sponsor and CMO become more comfortable with one another. Just remember to document such changes and explain why they happen.
Recalls: Because product recalls affect the reputations of both sponsor and manufacturer, none should be implemented without knowledge and input of the CMO — especially if the reasoning points to a manufacturing problem. You will probably need that company’s help during investigation and/or execution of a recall.
Change control is such a major issue that the FDA included it in the guidance. You can give a CMO latitude for certain types of process change, but license-reportable changes require the product sponsor’s involvement. Figure 3 illustrates a scenario that works. Notice the many touch points involved as well as the need for effective communication. Change can come from both sides, so be cooperative.
Person in the Plant: Having a representative of the sponsor company can either add value or cause problems for the CMO. Define the roles and responsibilities, what that person can and cannot do. Some things need approval from your company’s headquarters, so build that into the “person-in-plant” expectations. A well-managed relationship between the CMO and that person can add tremendous value to a project. He or she gains credibility at the CMO and works on its schedule rather than phoning in from halfway around the world. On the sponsor side, it provides a good connection into the CMO.
Auditing: Besides routine GMP auditing every year or so, a quality agreement should include “for-cause” audits. Such causes could include poor production success, a rash of repeat deviations, a recall, and poor inspection or routine audit results. If the CMO’s quality or production head is fired, for example, or its organization is restructured, then a for-cause audit may be triggered as well. That could suggest that something is happening at the plant that you do not know about.
Inspections: Make sure the CMO informs you of all impending or sudden inspections. Be prepared to be present at that location and available should a product-related question arise. But remember: This is an inspection of the CMO’s facility, so don’t try to take charge. A cooperative relationship will be most effective. It’s better to sit in a conference room for a week doing other work and still be available if a product question comes up than have to fly to the location after everything falls apart. Negotiate for the right to be there for inspections, but don’t get pushy.
Filings: You need the CMO’s help with regulatory filings. At the new scale (once a process has been scaled up), the CMO staff are the experts. Define your expectations and negotiate them.
Validation: In general, I leave plant-related validations to the CMO and audit the results. But I should be actively involved in product-specific validations (e.g., related to analytical methods and manufacturing processes). I review and approve protocols and reports. For equipment cleaning, I review and approve the definitions of clean but allow the CMO team to manage associated validations — only reviewing and ultimately auditing the end result, myself. Any changes to the clean definition require sponsor approval.
Annual Product Review (APR): You need to take charge of the APR, but you will need your CMO’s help — and data. This approach presents a good time for both parties to meet. I usually make it a face-to-face meeting, which helps to develop team bonding and cooperation. The CMO can and usually will have good ideas for improvements that can benefit both parties. An effective and successful APR can be a key part of continuous improvement, so use it effectively.
Dispute Resolution: The “You did not do what the contract says” type of dispute resolution is for lawyers to work out. But if sponsor and CMO do not agree on a decision or pathway forward, then the project teams can negotiate a resolution. This may involve an independent laboratory for testing issues or require executive management’s involvement for other matters. Define a clear pathway in your quality agreement.
Addendum: I advocate concluding with all items that can change often but have no real effect on the outsourcing relationship (e.g., identifying contact people and providing their email addresses, phone numbers, and so on). You may think of other types of information to include here — for which you won’t need to ask permission to make changes. Again, define that up front in the opening sections.
Be Prepared
This list is not complete. You can’t simply turn it into a checklist and never have to revisit content again. Many quality agreements between CMOs and sponsors might cover all these elements; not all will require all these points, and some will require more. We put these agreements in place to make our business relationships easier, not to satisfy regulators or arbitrary requirements.
Once a good quality agreement has been negotiated, the parties involved won’t necessarily follow it unless it is enforced. Spend time working out the details. Then put those into action. I cannot guarantee everything will be fine, but your chances are better with a good agreement than without one. Without these early negotiations and documentation, how can either party know what the other’s expectations are? You don’t want to work these things out during an unfolding crisis.
Author Details
Peter Calcott is president of Calcott Consulting LLC; 931 Mendocino Avenue, Berkeley, CA 94707; 1-510-585-8256; peterc@calcott-consulting.com.
Reference
1.) CBER/CDER/CVM. 2013, May. Guidance for Industry: Contract Manufacturing Arrangements for Drugs — Quality Agreements. US Food and Drug Administration, Rockville, MD. www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM353925.pdf.