Assurance that monoclonal antibody (MAb) therapeutics produced using large-scale animal cell culture are free of adventitious agents is based on three complementary approaches. First, a master cell bank is extensively characterized to demonstrate its freedom from adventitious agents. In addition, end-of-production cell culture material is tested and demonstrated to be free of adventitious contaminants. Finally, the viral clearance capacity of a purification process is assessed using model viruses.
PRODUCT FOCUS: MONOCLONAL ANTIBODIES
PROCESS FOCUS: PURIFICATION
WHO SHOULD READ: MANUFACTURING, PROCESS DEVELOPMENT, REGULATORY AFFAIRS, AND PROJECT MANAGERS
KEYWORDS: MMV, XMULV, CELL CULTURE, CHO CELLS, CENTRIFUGATION, VIRAL CLEARANCE, CGMP
LEVEL: INTERMEDIATE
Model viruses are used to determine the viral clearance capacity of purification process unit operations. Xenotropic murine leukemia virus (x-MuLV) and mouse minute virus (MMV) are commonly used during validation of the viral clearance potential for various purification process unit operations as required by regulatory agencies (1). Model viruses are useful because data generated during evaluation of the unit operations in a MAb purification process can be generally extrapolated to other viruses with similar physicochemical characteristics.
Xenotropic murine leukemia virus, a retrovirus with relatively low resistance to physicochemical inactivation, is useful for understanding how related adventitious enveloped viruses would respond to clearance processes. For nonenveloped viruses, the same is true of MMV, a DNA virus with relatively high resistance to physicochemical inactivation. Virus stocks with higher titers are advantageous because they potentially allow for a greater log reduction claim in a given unit operation. The model virus titer should therefore be as stable as possible.
Presently, there is little to no information in the public domain regarding stability of x-MuLV or MMV in long-term storage. The study presented here is, to the best of our knowledge, the first of its kind. It has been written to provide information on these commonly used model viruses. Data regarding virus production and storage conditions described here have been used to determine a minimum appropriate timeframe in which stocks of MMV and x-MuLV maintain a stable titer in cold storage. We use a standard tissue culture infectious dose assay (TCID50) to reassess the titer of MMV and x-MuLV stocks at one year or more after production.
Relying on an indicator cell line that exhibits cytopathic effects (CPE) after a specified incubation period, cell-based infectivity assays such as TCID50 are the most commonly used methods for virus quantification. The TCID50 assay has an accepted variability of ±1.0 log10 TCID50/mL. For x-MuLV, the most commonly used indicator cell line for these assays is PG4 (a feline astrocyte cell line), and CPE is typically assessed on day seven after inoculation. The x-MuLV–induced CPE on PG4 cells is characterized by foci (formed by loss of contact inhibition and growth of cells) piled on top of a contact-inhibited cell monolayer. For MMV, the most commonly used indicator cell line in these assays is an SV-40 large T-antigen–transformed human newborn kidney cell line known as 324K (2), and CPE is typically assessed on day 10 after inoculation. The MMV-induced CPE on such cells is characterized by extensive cell death (where virus concentration is high) and abnormal cellular morphology such as pseudopod formation and vacuolization.
Our studies summarized here assess the potential impact of several factors on long-term stability for each of those high-titer model viruses.
Materials and Methods: x-MuLV Stability TestingProduction of x-MuLV Virus Stocks: Mv1Lu cells infected with x-MuLV were grown to confluency in a sufficient number of T-175 flasks using Mv1Lu maintenance media: Dulbecco’s modified Eagle medium (DMEM) without phenol red, supplemented by 10% fetal bovine serum (FBS), 1% L-glutamine (L-glut), and 1% sodium pyruvate (NaPyr). Originally, cells were obtained from the American Tissue Culture Collection (ATCC# CCL-64), and the infected cell line was established in-house using an x-MuLV stock from BioReliance (www.bioreliance.com, lot number XML080902P). Cells were then washed with 1× Dulbecco’s phosphate-buffered saline (1× DPBS) and trypsinized with 0.05% trypsin–EDTA either with or without phenol red. If trypsin with phenol red was used, we spun the cells at 230g for 10 minutes before resuspending them in x-MuLV production media at the same volume. Production media consisted of Mv1Lu maintenance media supplemented at 1% with penicillin–streptomycin–amphotericin B (PSA).

We used a standard hemocytometer to count the cells once they were pooled, then seeded the cells into the required number of 10-chamber cell factories from Corning Life Sciences (www.corning.com/lifesciences) at 1.5 × 104 cells/cm2 using ∼1.5 L of x-MuLV production media per cell factory. We used a 10-chamber CellSTACK system to grow adherent cells. Cell factories were incubated at 37 °C in 5% CO2 for four days. Once monolayer confluencies reached 100% on day four, cell factories were harvested, fed again with x-MuLV production media, and returned to the incubator. Harvesting was done again on days six and eight. Virus from the day-eight harvest ultimately would be concentrated using a Cogent M TFF system from Millipore Corporation (www.millipore.com).
Each harvest day represented one seed bank. All harvests were spun at separately at 9,000g for 30–60 min, then the clarified supernatant was poured off in the direction of the pellet. We concentrated viruses from the clarified supernatant using either an ultracentrifuge, a hollow-fiber TFF system, or the Millipore Cogent TFF system. Original x-MuLV titrations took place within one to two weeks of vialing, and only lots created with ultracentrifugation or the hollow-fiber system were used for titer stability assessment. We used lots created with the Millipore system to compare the efficacy and efficiency of hollow fibers with that of flat sheet fibers for the purpose of virus concentration, referring to ultracentrifugation as a benchmark.
Ultracentrifugation: We concentrated x-MuLV from clarified supernatant using one of two ultracentrifugation
methods: either centrifugation at 9,000g for 24 hours or at 53,000g for an hour and 45 min. After each spin cycle, supernatant was poured off in the direction of the pellets and replaced with fresh clarified supernatant poured in at the opposite side of the pellets. After the final concentration cycle, supernatant was poured off, and pellets were allowed to soak overnight in BTNE buffer (67 mL 7.5% BSA, 10 mL 1-M Tris, 5.84 g NaCl, and 0.38 g EDTA brought up to 1,000 mL with tissue-culture–grade water) or TNE (BTNE without BSA) at a total volume of ∼1/150 the original cell factory seed volume (usually 100 mL and 15,000 mL, respectively). Resuspended viruses were vialed the next day at 1.0 mL/vial and stored at −70 °C or in vapor-phase liquid nitrogen (LN2).
Tangential-Flow Filtration (TFF) uses a porous barrier to separate suspended or dissolved materials based on their size or molecular weight, eliminating the need for centrifugation, solvent-phase changes, or other product-damaging techniques. We tested this method as an alternative to ultracentrifugation because it is faster and perhaps less harmful for concentrating x-MuLV. The entire clarified supernatant (10 × 10-chamber cell factories) can be processed within one day using TFF, whereas ultracentrifugation alone can require up to four or five days.
We used either a hollow-fiber MiniKros TFF system (Spectrum Laboratories, Inc., www.spectrapor.com) constructed in-house or the Cogent system to concentrate x-MuLV. The latter employs flat-sheet fibers rather than the hollow fibers of the former. The Cogent system uses a diaphragm pump, its main feed pump, to recirculate retentate. Application of a positive or negative pressure differential across the selectively permeable barrier drives sieve-like separation. Smaller constituents pass through the barrier with the solvent as filtrate, whereas the larger solutes (such as x-MuLV) are retained.
Hollow-Fiber System: Once assembled, the fiber membrane was wetted by pumping 30% isopropanol (IPA) into the module, which was subsequently flushed with suitable quality water followed by PBS solution. Clarified supernatant was poured into the product reservoir, and a slight resistance was applied to the output end of the hollow-fiber module. Supernatant was thus circulated through the system until a desired volume or concentration of product remained in the product reservoir. What remained in the fiber membrane was drained by reversing the pump direction. Supernatant remaining in the concentrator after pump reversal was poured into a sterile container and subjected to ultracentrifugation as described above. Concentrated product was vialed at 1.0 mL/vial and stored at −70 °C or in LN2 with either BTNE or TNE storage buffer.
Flat-Fiber System: We set up our Cogent M system according to the manufacturer’s recommended procedures. For these studies we chose a Pellicon 2 cassette (P2 Biomax 500-K C screen) because it most closely matches the current MiniKros module. To remove the packaging, the membrane was flushed with deionized water to the recommended throughput. We used an input pressure of 18 psi, and the actual output pressure during the UF process was 16 psi. The flow rate used was 750 mL/min (7.5 L/min/m2), and the total system hold-up volume was 200 mL. The starting volume of the feed was estimated to be 7.2 L. This includes all residual deionized water used to flush the membrane and system. The feed was about 7.2 L of day eight harvest from our x-MuLV production. We froze conditioned medium at −30 °C on 9 February 2007 and thawed it at room temperature (RT) in a hood overnight for the TFF process performed on 26 February 2007.
After processing, the product was recovered by a low-point drain built into the Cogent system and a 100-mL 1× DPBS flush. After recovery, the total volume of retentate was brought to 350 mL with the buffer-flushed material for a total concentration factor of 20×. We overconcentrated the ultrafilter to place a buffer flush step that could increase product recovery. The membrane was cleaned with 1-N NaOH at room temperature at a flux excursion of 10 psi for 30 minutes. The Pellicon cassette and tubing were discarded after use.
Materials and Methods: MMV Stability TestingProduction of MMV Virus Stocks: NRK-52E cells (ATCC #CRL-1571) were grown to confluency in a sufficient number of Corning T-175 flasks using DMEM supplemented with 5% FBS, 1% L-glut, and 1% NEAA. The cells were rinsed with 1× DPBS and trypsinized using 0.05% Trypsin–EDTA, either with or without phenol red. If trypsin with phenol red was used, we spun the cells at 230g for 10 minutes before resuspending them in media at the same volume with MMV production media composed of DMEM (without phenol red) containing 5% FBS, 1% L-glut, 1% NEAA, and 1% PSA. With a standard hemocytometer we counted the cells once they were pooled. We seeded the cells into the required number of 10-chamber cell factories and one single-chamber cell factory at 1.5 × 104 cells/cm2 using 1.5 L of MMV production media for each 10-chamber factory and 150 mL for the single-chamber one.
The following day, we inspected cells for the target monolayer confluency of 20–50%. Those from the single-chamber cell stack were trypsinized, and a viable cell count was obtained for estimating the total number of cells in each 10-chamber cell factory. Based on that estimation, we calculated a target multiplicity of infection (MOI, the ratio of infectious virions to cells) of 0.01.
To prepare the cell factories for inoculation, we first decanted off the production media and then rinsed each factory with 200 mL of 1× DPBS. A virus inoculum was prepared in 200 mL of production media and added to each cell factory. The factories were placed in a 37 °C incubator with 5% CO2 incubator for 60–90 minutes to allow for virus adsorption. After that time had elapsed, 1,300 mL more MMV production media was added and factories were returned to the incubator. They were observed until the cell monolayers showed 90–100% CPE (generally four or five days), at which point three consecutive freeze–thaw cycles were performed to harvest MMV.

Our virus harvest was clarified by centrifugation at 9,000g for 30–60 min. MMV was concentrated from the clarified supernatant by centrifugation at 100,000g for an hour and 45 minutes in a Beckman Coulter Type 45 Ti rotor. After each spin cycle, supernatant was poured off in the direction of the pellets and replaced with fresh clarified supernatant poured in at the opposite side. After the final concentration cycle, supernatant was poured off, and pellets were then allowed to soak overnight at 2–8 °C in TNE buffer (Tris, NaCl, and EDTA tetrasodium dihydrate) at a total volume equal to 0.066–0.100 the original cell factory volume. The following day, all suspensions were combined into one, and the pellets were sonicated for one to two minutes before 0.22-µm filtration. Filtered product was aliquotted and stored at −70 °C or in vapor-phase LN2. Stocks were then retitrated at least one year after their initial titer determination.
Materials and Methods: MMV Centrifugation AnalysisNRK cells in a 10-chamber cell factory were infected, harvested, and clarified as described above. To assess the impact on the titers of stocks created from these manipulations, we varied relative centrifugal force (RCF), concentration time, and resuspe
nsion temperature in the processing of clarified supernatant. The rotor we used in all conditions was the Beckman Coulter Type 45 Ti rotor. We compared titers of the stocks from conditions 1–5) with those of three stocks concentrated at a maximum RCF of 100,000g.
Set 1, Varying RCF and Centrifugation Time: Condition 1 was a control wherein the virus suspension was concentrated according to the standard MMV protocol described above. We ran condition 2 to determine whether an additional 49,000g of maximum force would significantly increase the titer of MMV stock. Similarly, we ran condition 3 to determine whether centrifugation at a lower RCF for a longer period than that described in the standard protocol would significantly increase the MMV titer. Here are the conditions:
Condition 1: clarified supernatant spun at 29,300 RPM (maximum RCF of 100,000g, average 67,300g) for an hour and 45 minutes
Condition 2: clarified supernatant spun at 35,800 RPM (maximum RCF of 149,000g, average 100,000g) for an hour and 45 minutes
Condition 3: clarified supernatant spun at 26,700 RPM (maximum RCF of 83,000g, average 55,800g) for 18 hours.
After each spin cycle, supernatant was poured off in the direction of the pellets. Then 5 mL of TNE buffer (30 mL total per cycle, or 1/10 the original volume of concentrated clarified supernatant) was put into each centrifugation tube, all angled so the buffer covered the pellets. We let the tubes sit overnight at 2–8 °C. The following day, virus suspensions from each respective test condition were combined and sonicated for one to two minutes before 0.22-µm filtration. The filtered product was vialed at 1.0 mL/vial and stored at −70 °C.
Set 2, Varying RCF and Resuspension Temperature: Test set 2 was run to determine whether increased RCF significantly condensed the virus pellets so that resuspension would be hindered, and whether resuspension at physiologic temperature had any apparent impact on the final certified titer. Viruses were concentrated at an RCF of 149,000g in conditions 4 and 5. Condition 4, wherein viruses were resuspended using the standard overnight incubation at 2–8 °C, served as a control for condition 5, wherein virus was resuspended overnight at 37 °C on a shaker platform. As in set 1, the supernatant was poured off before each virus pellet was resuspended in 5 mL of TNE (15 mL total per temperature). We allowed the pellets to resuspend overnight, and the next day combined them, sonicated for 1–2 minutes, and then ran a 0.22-µm filtration. The filtered product was then vialed at 1.0 mL/vial and stored at −70 °C.
Condition 4: clarified supernatant spun at 35,800 RPM (maximum RCF of 149,000g, average RCF of 100,000g) for an hour and 45 minutes, then virus resuspended overnight at 2–8 °C
Condition 5: clarified supernatant spun at 35,800 RPM (maximum RCF of 149,000g, average 100,000g) for an hour and 45 minutes, then virus resuspended overnight at 37 °C in 5% CO2 on a rotating shaker platform set at 150 RPM.
Materials and Methods: Virus Titer DeterminationFor x-MuLV titer determination, we cultured PG-4 cells (ATCC# CRL-2032) in Corning T-175 flasks using McCoy’s 5A medium supplemented with 10% FBS and 1% L-glut. After three to four days, once full confluency was reached, the cells were washed with 1× DPBS. Then we trypsinized them off the flask using 0.05% Trypsin–EDTA with phenol red and resuspended them in seed media containing McCoy’s 5A, 2% FBS, 1% L-glut, and 1% PSA. We determined a viable cell count using a standard hemocytometer and seeded the cell suspension into 96-well plates (Corning) at a density of 2.1 × 104 cells/mL and 150 µL of cell suspension per well. The cells were returned to a 37 °C, 5% CO2 incubator overnight to allow their adherence on the bottom of the wells.
For MMV titer determination, we cultured 324K cells (from Peter Tattersall at Yale University) in Corning T-175 flasks using DMEM supplemented with 10% FBS, 1% L-glut, and 1% NEAA. After three to four days, once full confluency was reached, the cells were washed with 1× DPBS. Then we trypsinized them off the flask using 0.05% Trypsin–EDTA and resuspended them in seeding media containing EMEM and 2% FBS or DMEM, 2% FBS, 1% L-glut, 1% NEAA, and 1% PSA. We made a viable cell count using a standard hemocytometer, then seeded the cell suspension into 96-well plates (Corning) at a density of 1.7 × 104 to 2.1 × 104 cells/mL with 150 µL of cell suspension per well. The cells were returned to a 37 °C, 5% CO2 incubator overnight to allow for their adherence to the bottom of the wells.
The day after seeding, we inspected the wells under a microscope for health, freedom from microbial contamination, and the desired cell confluency of 20–50%. Neat virus preparation was subjected to nine serial 10-fold dilutions in 1.5-mL Eppendorf tubes using seed media as the diluent (0.1 mL of virus suspension was added to 0.9 mL of media for each dilution). Each dilution was inoculated onto one column (eight wells) of a 96-well plate at 50 µL/well, starting with the highest dilution (10−9). Negative control wells were inoculated with the same media used for virus dilutions.
Plates were returned to a 37 °C, 5% CO2 incubator for seven days before virus-induced CPE observation. On day seven for x-MuLV and day 10 for MMV, we scrutinized all inoculated wells for CPE. We calculated TCID50 titer using the common Spearman–Kärber method. For virus stocks used in titer reassessments, most initial titration determinations took place one to two weeks after vialing; retitrations took place after a year or longer.
Results and DiscussionVirus lot numbers in the tabular data represent the date of vialing (MM/DD/YY format). The titers of all replicate titrations for a given virus lot were within 1.0 log10 TCID50/mL of each other. Tables 1 and 2 show how the original and new x-MuLV titers vary by concentration method and storage buffer, respectively. Table 3 compares x-MuLV titers obtained using ultracentrifugation and flat-sheet fiber TFF. Table 4 shows the original and new MMV titers. Tables 5–7 show the results of MMV centrifugation analysis. All titer values are shown in log10 TCID50/mL format at a 95% confidence interval.
Table 1: x-MuLV titer comparison

Table 2: x-MuLV virus titers (comparing ultracentrifugation and Cogent M TFF system); all virus lots stored in vapor-phase LN2
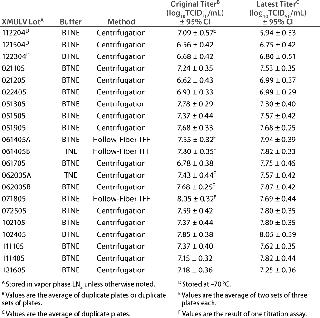
Table 3: MMV titer comparison

Table 4: Set 1, varying RCF and centrifugation time

Table 5: Set 2, varying RCF and resuspension temperature

Table 6: Negative control MMV stock titers

x-MuLV Stability Testing — Temperature: Tables 1 and 2 show the results of the original titrations and retitrations for all x-MuLV stocks tested for stability. Results indicate that storage of x-MuLV stocks in LN2 may provide more viral stability than at −70 °C. Retitration data show that no lots stored at −70 °C meet our minimum titer for viral clearance applications. Among the three lots stored at −70 °C, x-MuLV 112204 fell in titer by more than 1.0 log10 TCID50/mL from its initial titration, but it is uncertain whether that change can be directly attributed to temperature. That result is in contrast with the other two lots, x-MuLV 121504 and x-MuLV 122304, for which retitration showed less than 0.3 log units of difference from the original certified titers, suggesting that long-term storage at the same temperature did not adversely affect their stability. We need to test more stocks stored at −70 °C to conclusively rule out this storage temperature.
On the other hand, all 18 stocks of x-MuLV stored in vapor-phase LN2 remained stable within the assay variability of ±1.0 log10 TCID50/mL. Only two lots gave titers below 7.0 log10 TCID50/mL in both their original and repeat titrations. Given that there are relatively few replicate data points regarding storage of x-MuLV at −70 °C, it is recommended that stocks of x-MuLV be stored in LN2 to provide the greatest assurance of long-term virus stock stability.
Concentration Method: Titers of x-MuLV stocks prepared using the hollow-fiber module were comparable to those prepared by the centrifugation method or with the Cogent system. Lots XMULV 061405A, XMULV 061405B, and XMULV 071805 all were produced using a hollow-fiber module. After storage in LN2, those lots were retitrated, and all are within 0.4 log units from the original values (Tables 1 and 2). Those values are on the same order of magnitude as the lots prepared using ultracentrifugation. TFF is equally effective, less time-consuming, and potentially less damaging. So it is a viable replacement of (or addition to) ultracentrifugation for x-MuLV stock production. And as Table 3 demonstrates, concentration with flat-sheet fibers yields titers similar to those achieved with the MiniKros module or with ultracentrifugation using TNE or BTNE as a storage buffer.
Bovine Serum Albumin: The effects of BSA and different purification methods were assessed for their impact on final virus titer. BSA is an abundant serum protein, and its main purpose is to regulate the colloidal osmotic pressure in blood. It also has a good binding capacity for water, Ca2+, Na+, K+, fatty acids, hormones, bilirubin, and drugs. Because of its ability to bind a wide range of ions and molecules, BSA was believed to help stabilize virus particles during storage and thawing and was included as a component of storage buffer.
To test that hypothesis, we prepared two sets of lots and stored them in buffer with and without BSA. The first set, prepared using hollow-fiber TFF, included lots XMULV 061405A (BTNE) and XMULV 061405B (TNE). The second set, prepared using ultracentrifugation, included lots XMULV-TNE and XMULV 062005 (BTNE). Both virus lots within each set were vialed on the same day. These lots showed no significant difference in titer after long-term storage (Table 1), regardless of the presence of BSA. All lots produced using TNE, including those made with the Cogent system, showed titers that were comparable to those prepared with BTNE. These data indicate that BSA is not a necessary virus storage buffer component because BTNE does not seem to provide better stability than TNE alone. Since BSA does not seem to serve any practical purpose, it is therefore recommended that its use in x-MuLV storage buffer be discontinued as part of efforts to minimize or eliminate the use of animal-sourced components.
Regardless of the parameter tested, nearly all lots were well within the accepted variability of the TCID50 assay. One limitation of this study was that more lots of x-MuLV were stored in vapor phase LN2 than at −70 °C, but we believe that vapor-phase LN2 is preferable to −7
0 °C for storing x-MuLV lots. All lots of x-MuLV were titrated about two years after vialing of the oldest three lots. Because a significant drop in titer was seen in only in the oldest lot, which was stored in −70 °C, we believe that the actual shelf life of x-MuLV banks stored in vapor-phase LN2 is a minimum of two years and perhaps longer.
MMV Stability Testing: We retitrated a set of MMV stocks to assess stability of virus titers in long-long term storage (Table 3. Of the nine stocks tested, none showed a significant change in titer; their latest titers were within ±1.0 log10 TCID50/mL from the originals. Given that the titer of the oldest lot, MMV 070804, was stable at its retitration (∼2.5 years after original titration), we estimated that the minimum shelf life of MMV is three years.
MMV Centrifugation Analysis: As with x-MuLV, we altered the production method of MMV to determine the impact of such changes on the final stock titer. We varied centrifugation force and time as well as resuspension temperature postconcentration.
Table 4 shows the results of test set 1 (RCF and centrifugation time). In general, there seems to be no significant benefit to concentrating MMV at the higher RCF for the same period as in the standard protocol. The same conclusion applies to concentrating MMV at a lower RCF for a longer time. The difference in titer between the average of the three standard MMV stocks (Table 6) and those in set 1 was less than 1.0 log10 TCID50/mL for all conditions, and the greatest difference was 0.27 log10 TCID50/mL. Moreover, titers obtained in set 1 are all within 1.0 log10TCID50/mL of one another.
Similarly, titers obtained in set 2 (RCF and resuspension temperature, (Table 5) showed no significant difference from the average of the three negative control stocks. It should be noted, however, that the highest titer obtained among all virus lots tested was from the stock spun at 149,000g maximum and resuspended at 37 °C (condition 5, Table 5). That challenges a 1974 paper by Harris, et al. claiming that storage of MMV at 35 °C for 220 minutes could lower viral titer by 1.0 log10 TCID50/mL (3). Our result also acknowledges the possibility that some virus pellet remains stuck to the side of the centrifugation tube and were released by shaking and the 37 °C resuspension temperature. Considering these results, it is worth replicating condition 5 with conditioned media from one 10-chamber cell stack (∼1,500 mL) to discover whether or not the size of the virus stock being concentrated affects the final stock titer.