In the arsenal of biophysical techniques available for rapidly monitoring the stability of protein formulations, spectroscopic techniques have some convincing advantages over others (1, 2). The main advantages to using methods such as circular dichroism (CD), infrared spectroscopy (IR) and fluorescence spectroscopy are their extremely high sensitivity (favorable signal-to-noise ratios), freedom from sample interactions with column resins or extrinsic probes (noninvasive techniques), and coverage of an extremely broad protein concentration range — from pM to mM (3, 4).
To reduce biopharmaceutical development times and costs, different analytical techniques and experimental protocols are applied for rapid screening of formulations toward finding appropriate stability
PRODUCT FOCUS: MONOCLONAL ANTIBODIES (MABS)
PROCESS FOCUS: PREFORMULATION AND FORMULATIONS ANALYSIS, DOWNSTREAM PROCESS OPTIMIZATION
WHO SHOULD READ: QA/QC, PROCESS DEVELOPMENT, FORMULATORS
KEYWORDS: STABILITY, DENATURATION, FLUOROPHORES, CIRCULAR DICHROISM, FLUORESCENCE SPECTROSCOPY, BIOPHYSICS
LEVEL: INTERMEDIATE
conditions. Therefore, accelerated stability screening studies using biophysical techniques are often early activities in preformulation (5, 6). Such studies provide information about the thermal stability of protein formulations varying by, for example, pH, ionic strength, and ingredient concentration. Techniques routinely applied for preformulation stability studies include differential scanning calorimetry (DSC); Fourier-transform infrared (FTIR), ultraviolet/visible (UV/vis), or CD-spectroscopy; and/or light-scattering techniques (static, dynamic) (6,7,8).
Here we focus on fluorescence spectroscopy as an alternative for early formulation development because
-
tryptophan fluorescence is very sensitive to protein conformational changes and provides information about changes in secondary and/or tertiary structure
-
only a low amount of protein (∼50 µg per sample or less) is required for analysis
-
only minor chemometrical efforts are required to quickly derive relevant stability indicating data
-
data obtained from fluorescence measurements are usually quite robust.
Many papers describe the potential of intrinsic tryptophan fluorescence spectroscopy in protein chemistry (9). However, fluorescence spectroscopy is not so common in pharmaceutical formulation sciences. A number of papers have described the use of fluorescence intensity or maximum emission wavelength (λmax) to monitor conformational changes in proteins. Famous examples include conformational studies focusing on nuclease A from Staphylococcus aureus as a model protein, a single-tryptophan protein that displays essentially two-state thermodynamics upon thermal unfolding, which can be monitored easily using fluorescence spectroscopy (10,11,12,13). A simulation study showed that steady-state fluorescence intensity is directly related to the mole fraction of protein states (unfolded/folded) and can therefore be used to monitor unfolding transitions in proteins (14). Recent pharmacological and biophysical studies used fluorescence data to monitor the thermal stability of proteins such as monoclonal antibodies (15), recombinant vaccines (16), human interferon-α2a (17), α-amylases (18, 19), and chymotrypsin inhibitor (20), to name just a few.

WWW.BOEHRINGER-INGELHEIM.COM
What makes tryptophan such an attractive fluorescence probe is the huge change of dipole moment of its indole side chain upon excitation at 295–300 nm (9, 21, 22). This excited electronic state (also referred to as the 1La state) responds very sensitively to conformational changes in the local microenvironment, resulting in a fluorescence maximum shift and changes in fluorescence intensity, anisotropy, lifetime, and so on. Although these processes are not entirely clarified, semiquantitative quantum-mechanical and molecular-mechanical approaches have gradually contributed to understanding the nature of those electrostatic interactions (9, 23,24,25).
Quenching is another mechanism affecting the fluorescence properties of proteins. Many studies have been performed using external quenchers such as acrylamide or iodide to localize surface-exposed, accessible, or buried tryptophan residues within a protein structure. Quenching effects can also arise intramolecularly from various tryptophan-neighboring amino acid residues such as cysteine/cystine, histidine, tyrosine, lysine, glutamine, asparagine, and glutamic and aspartic acids (26). The change of fluorescence intensity upon thermal denaturation depends strongly on the microenvironments of the tryptophan residues and can, in principle, be predicted — if a protein’s structure is known. In most cases, however, the structure of a novel therapeutic protein is not known with atomic precision. Even so, fluorescence spectroscopy can provide very useful and comprehensive data about protein stability.
Folding–unfolding equilibria of small globular proteins often can be described easily assuming a two-state model as long as only one experimental parameter is varied — e.g., pH, temperature, or the concentration of denaturant (27). The situation becomes more complex when a protein is structured into multiple domains that do not unfold cooperatively, as with immunoglobulins (28,29,30), because satisfactory models do not exist for fitting their experimental data.
Our aim was to expand fluorescence spectroscopy to a universal tool for preformulation of tryptophan-containing proteins and show the robustness of this technique that allows rapid screening and characterization of different formulations. A possible experimental application could be the study of pH-dependent thermal transitions in proteins. The technique also offers the possibility of extending an intrinsic fluorescence approach to investigating the influence of ionic strength, detergents, or other ingredients on a protein’s structure in a given formulation. We found that fluorescence spectroscopy fully meets these requirements and thus proves a valuable tool in the preformulation development of proteinaceous biopharmaceuticals.
Materials and MethodsAmino Acids: The amino acids L-tryptophan, L-tyrosine, and L-phenylalanine were purchased from Sigma-Aldrich (www.sigmaaldrich.com). Unless otherwise specified, their fluorescence spectra were recorded from solutions with a final concentration of 10 nM at 25 °C in water.
Monoclonal Antibody: Boehringer Ingelheim Pharma GmbH & Co. KG (www.boehringer-ingelheim.de) provided a humanized monoclonal antibody (IgG). Using a Spectra/Po
r 7 instrument from Spectrum Laboratories (www.spectrapor.com), we aliquoted and dialyzed the initial aqueous antibody solution against assay buffers described below. Finally, the protein concentration was adjusted to 0.1 mg/mL and degassed immediately using a ThermoVac instrument from MicroCal LLC (www.microcal.com) before each measurement.
Fluorescence Spectroscopy was performed using an Aminco-Bowman Series 2 luminescence spectrometer from Thermo Electron Corporation (www.thermo.com) equipped with a self-designed and self-manufactured Peltier-controlled cuvette holder. We recorded tryptophan emission spectra at 310–390 nm upon excitation at 295 nm in steps of 2K over a 30–50 °C temperature range and in 1K steps over the 50–80 °C range. Before measurement, sample cells were equilibrated for 90 seconds at each temperature. After smoothing the raw spectra with a seven-point Savitzky-Golay algorithm, we plotted the intensities of the emission maxima and the position of the peak maxima (expressed as a ratio of emission intensity at 330 nm to that at 350 nm) against the temperature (31). Resulting thermograms were smoothed likewise with a seven-point Savitzky-Golay algorithm, and we obtained transition temperatures from where the abscissa was crossed by the first derivative of maximum-intensity over temperature and also the second derivative of the peak-position over temperature plot.
Differential Scanning Calorimetry (DSC) was performed using a VPDSC from MicroCal. We set the heating scan to 1 K/min and ran the experiment in the temperature 10–95 °C interval. Protein concentration was ∼1 mg/mL, and a reference cell was filled with the corresponding buffer. We analyzed heat capacity curves using Origin 7.0 software. The maximum of the heat capacity curve is defined as the denaturation temperature (Tm).
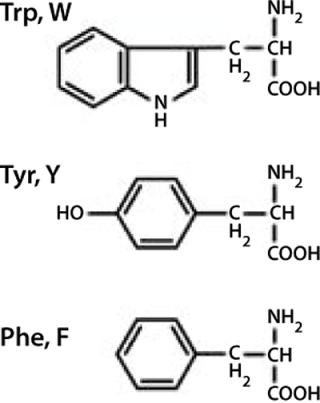
ResultsAs Figure 1 and Figure 2 show, proteins may contain three aromatic amino acids that show fluorescence in the near UV-range: tryptophan (Trp, W), tyrosine (Tyr, Y), and phenylalanine (Phe, F). Only two of those, tryptophan and tyrosine, contribute to the absorption maximum of proteins at 280 nm. To obtain fluorescence data predominantly from tryptophan residues, the excitation wavelengths range between 295 and 305 nm in most intrinsic fluorescence experiments.
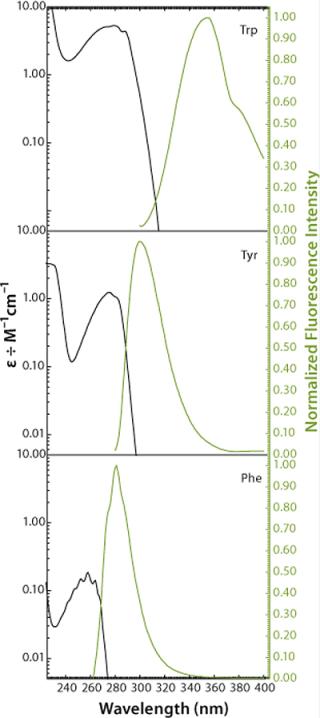
Performing fluorescence assays in that range has the further beneficial effect that mainly the 1La state of tryptophan (the state with the higher dipole moment) is excited, rather than the 1Lb state. This provides a much higher sensitivity of the fluorophore to changes in its microenvironment (21). Such changes are predominantly electrostatic and come from interactions between the fluorophore and adjacent groups. The effect can be observed in fluorescence emission spectra as a shift of fluorescence maximum resulting from relaxation and reorientation of the local field (24).
In apolar solvents such as 2-propanol the emission maximum of tryptophan can thus be found at 340 nm, whereas in polar solvents such as water that maximum lies at 355 nm (Figure 3). Influences of pH and temperature on the fluorescence of aqueous tryptophan have been studied in detail (32, 33). Although both have major effects on protein conformation, they have virtually no effect on the maximum emission wavelengths of free amino acids (Figure 4 and Figure 5). However, with increasing temperature, the fluorescence intensity decreases as a result of nonradiative processes such as intersystem crossing and photoionization competing with the fluorescence (32).
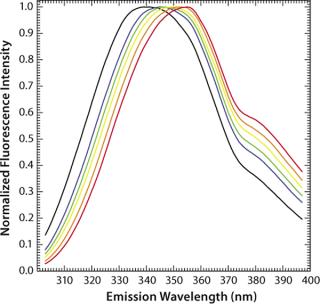
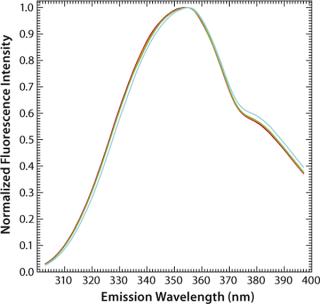
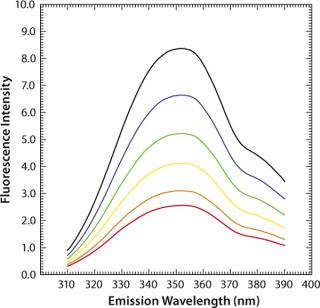
Another factor often critical to protein formulation is ionic strength, which has only a marginal (hardly any) effect on the fluorescence intensity and maximum emission wavelengths of aqueous tryptophan (Figure 6). The exact nature of these processes and their effects on fluorescence emission properties are important when investigating protein stability as a function of formulations in assays using temperature as a stress condition. It is crucial to understand the impacts of temperature, ion
ic strength, and such on fluorescence emission to be able to interpret correctly the observed data in an intrinsic fluorescence temperature-induced protein denaturation experiment.
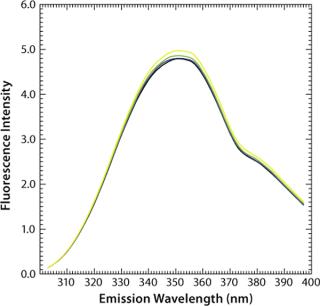
We used a monoclonal humanized antibody containing 20 tryptophans distributed over all antibody domains. Tryptophan residues that are fully or partially buried in the relative nonpolar interior of proteins have a fluorescence emission maximum in the range of 309 nm, as is found for azurin, whereas such residues that are less buried or are even in contact with polar interfaces exhibit fluorescence emission maximum above 350 nm.
MAb denaturation with guanidinium hydrochloride (GdnHC1) leads to a shift of λmax from 330 nm to 352 nm, indicating that all tryptophan residues become hydrated as a consequence of the loss of structural integrity (Figure 7). Concomitantly, fluorescence intensity increases almost by a factor of 1.5 (Figure 8). When such a protein is thermally denatured, λmax shifts from 330 nm to 338 nm, and its maximum intensity is decreased by a factor of 0.8, indicating that the denatured state obtained after denaturation are generally different from those resulting from thermal denaturation. In the temperature-induced denaturation experiments (Figure 8), strong, nonspecific protein–protein interactions seem to be stimulated, which brings tryptophan residues into a less polar environment than in GdnHCl-induced unfolding.
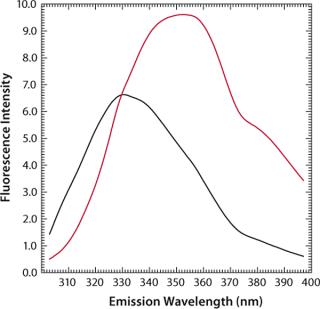
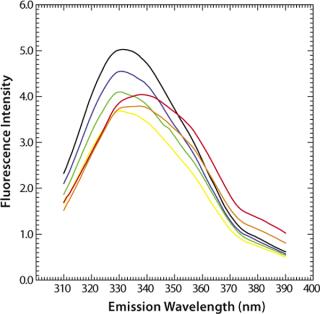
Figure 9 shows the pH-dependent course of fluorescence intensity and λmax of a MAb upon thermal denaturation. Starting around Fmax = 5 AU at 30 °C, the fluorescence intensity drops with increasing temperature to reach a minimum, Tm(Fmax), at 51.8 °C (pH 5.0, black curve in Figure 9) or at 65.1 °C (pH 7.0, green curve in Figure 9), as calculated from the first derivative of the averaged and smoothed intensity functions. With further increase in temperature, the fluorescence intensity rises, with a shoulder at 60 °C (pH 5.0), until it reaches a maximum at around 70 °C (pH 5.0) or 76 °C (pH 7.0).
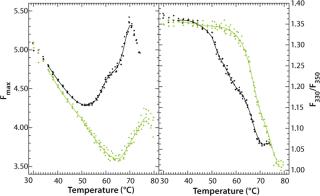
We could calculate that second major transition only with vague certainty because fluctuations appeared in the fluorescence emission spectra from light-scattering effects of aggregate formation. That effect must be considered by examination of such data. The ratio of the intensities at 330 nm and 350 nm (λmax) showed a major shift in Tm(F330/F350) toward higher wavelengths at 51.6 °C (pH 5.0) or 67.4 °C (pH 7.0), as calculated from the second derivatives of the F330/F350 ratio plotted against temperature (Figure 9B). For pH 5.0, both thermograms (Figure 9A and 9B) exhibit a shoulder between 58 °C and 65 °C, indicating a third transition upon thermal unfolding of the MAb.
Those two transition temperatures for Tm(Fmax) and Tm(F330/F350) were reproduced with high accuracy in three independent experiments, showing a standard deviation of <0.2 °C in all cases, and they correlate well with results obtained from DSC (Table 1). But it should be noted that, although these calculated transition temperatures appear to coincide, they do not necessarily underlie the same molecular mechanisms and should therefore be considered independently.
Table 1: Transition temperatures determined by monitoring the changes in the maximum fluorescence intensity (Fmax) and maximum emission wavelength (F330/F350) compared with data from differential scanning calorimetry (DSC).
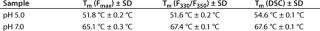
Our aim was to provide a quick, material-saving, and reliable analytical means to probe the thermal stability of monoclonal antibodies during early formulation development. To correctly evaluate and judge the measured fluorescence effects, the influence of stress and external factors (e.g., temperature and ionic
strength) on the fluorescence properties of an analyzed probe must be known. Therefore, we reinvestigated those applied stresses in preformulation development on the trypthophane fluorescence probe to evaluate tryptophan-protein fluorescence. We therefore reassessed intrinsic steady-state fluorescence of proteins because it allows detection of changes in protein conformation using as little as 0.5-mL samples at a concentration of 0.1 mg/mL or less for conventional IgG antibodies.
To monitor thermal denaturation, we used two observables: maximum fluorescence intensity and the wavelength of maximum emission (λmax) expressed as the ratio of the fluorescence intensities at 330 nm and 350 nm. Smoothing and differentiating the resulting thermograms with a seven-point Savitzky-Golay algorithm allowed us to detect three transition states indicated by a minimum, a shoulder, and a maximum in fluorescence intensity (Figure 9A). Furthermore, we calculated transition temperatures, Tm(Fmax) and Tm(F330/F350) (Figure 9A and 9B), with high reproducibility (standard deviations = 0.2 °C) and very good correlation with DSC results (Table 1).
The major limitation of steady-state fluorescence in multitryptophan proteins is that their fluorescence signal is always an average of all tryptophan residues in different local environments. Signal changes therefore cannot be resolved to molecular events unless structural information about a protein is available (e.g., from site-directed mutagenesis, X-ray crystallography, or nuclear magnetic resonance spectroscopy). An illustrative example elucidating the contribution of each tryptophan fluorophore to the overall fluorescence spectrum — as well as the influence of their local environments on their specific fluorescence properties — is found with barnase, the ribonuclease produced by Bacillus amyloliquefaciens (34). Loewenthal et al. convincingly demonstrated that fluorescence quenching by adjacent amino-acid residues (and resonance energy transfer to neighboring tryptophan residues), both contributing to the overall fluorescence spectrum of the protein in its native state (34). Consequently, unfolding of a multitryptophan protein can lead to an increase in the overall fluorescence intensity because of abrogation of local quenching or energy-transfer processes. It can also lead to a decrease because of novelly ascending intramolecular quenching processes or exposure to the aqueous phase (35). Both effects can, in principle, compensate for each other.
Initiating thermal denaturation of the MAb used in our work is characterized by an increase and then (after reaching a maximum) a decrease in fluorescence intensity, which indicates that, in principle, all the processes described herein can occur at distinct stages during thermal unfolding. By contrast, the maximum emission wavelength, λmax, shows an increasing red-shift upon denaturation (as with most proteins) because the tryptophan residues become more hydrated. This provides indispensable information that should be ascertained to obtain unambiguous results regarding thermal stability.
Furthermore, deviations of the λmax thermograms from a typical sigmoidal shape also indicate that thermal denaturation of the MAb is a multistate process (Figure 9, right panel). This is in accordance with earlier studies using DSC and demonstrating that two and more transitions can be observed because of partly uncooperative unfolding of individual antibody domains (28, 36, 37). But we find that, despite the complexity of protein structure and folding, steady-state fluorescence spectroscopy is a very economic and material-saving methodology providing consistent and easily accessible data on the thermal stability of monoclonal antibodies during preformulation development.
REFERENCES
ison with Theory. J. Phys. Chem. 94:3469-3479.