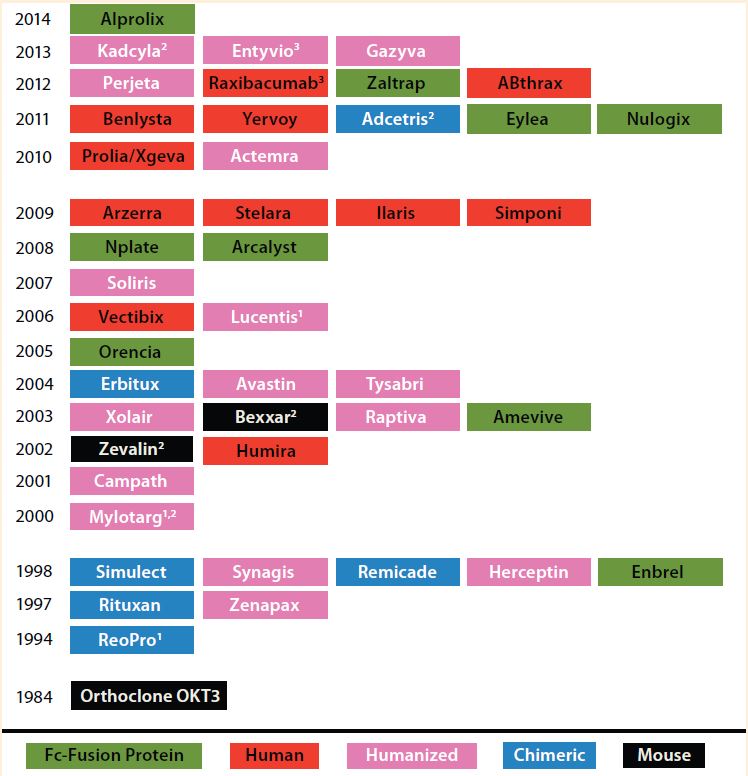
Table 1: Timeline for biotherapeutic MAbs and derivatives, including Fc fusion proteins, approved by the FDA in the indicated years; all are produced using recombinant DNA technology. The protein engineering technology on which each product is based is indicated. Nine of the 45 approved MAb products are Fc fusion proteins.
(1 First MAb approved under animal-efficacy rule 2 Antibody–drug conjugate 3 Fab/(Fab’)2 fragment)
Over the past three decades, 45 monoclonal antibody (MAbs) and MAb-derivative products have been approved for therapeutic use in the United States (Table 1). One class of antibody derivatives is growing in importance: Fc-fusion proteins.
Many biologically active proteins, including receptor ECDs (see “Abbreviations” box), cytokines, enzymes, and bioactive peptides have very short serum half lives because rapid renal clearance limits their exposure in target tissue (and, consequently, their pharmacological effect). The primary reason for fusing a biologically active protein with Fc is half-life extension (1).
| Abbreviations Used Herein
ADCC: antibody-dependent, cell-mediated cytotoxicity |
Since the first description in 1989 of a CD4-Fc fusion protein (2), interest in this class of engineered proteins has grown. The rationale for their development is that fusing a polypeptide — one that otherwise may not be a viable drug candidate because of its short in vivo half-life — to an immunoglobulin Fc converts that polypeptide into a practical therapeutic form. Thus, the ECD of the receptor CD4, which displays a very short in vivo half-life when expressed as a
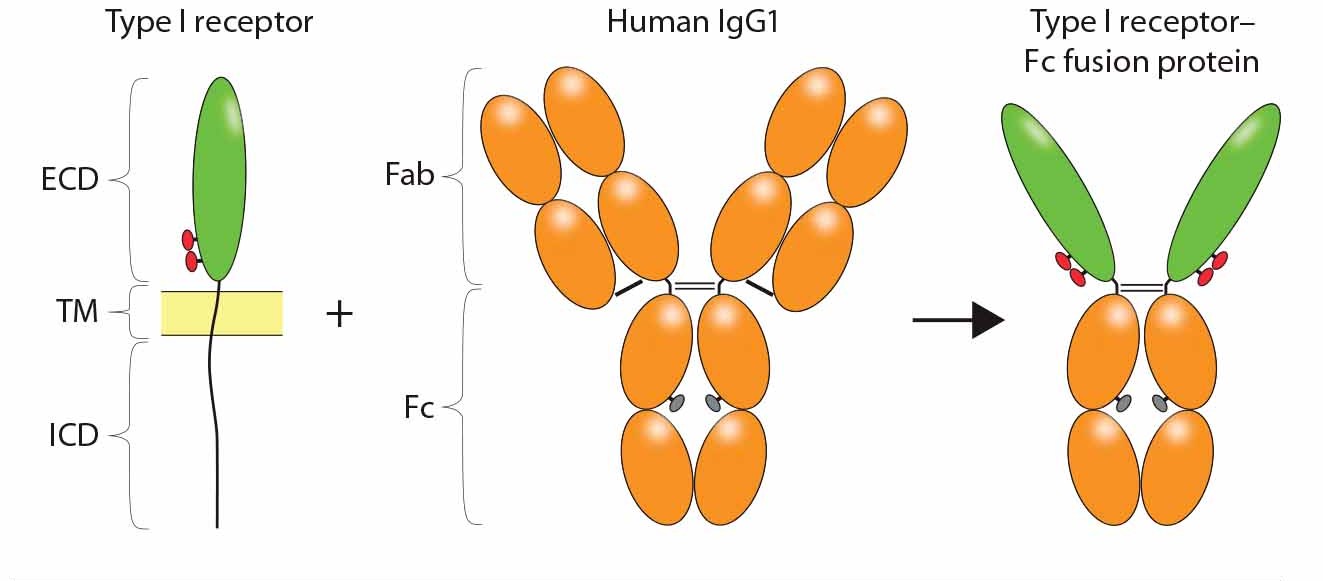
Figure 1: Construction of an Fc-fusion protein; typically, the ligand binding domains derived from the ECD of a Type I transmembrane receptor are combined with the Fc portion of a human IgG1 to form the Fc-fusion protein. Glycosylation sites encoded in the receptor domains (red ovals) or Fc domain (gray ovals) are indicated. Reproduced with permission (3).
soluble recombinant glycoprotein, is converted to a fusion molecule with an extended serum half-life.
Fc-fusion proteins are bioengineered polypeptides that join the crystallizable fragment (Fc) domain of an antibody with another biologically active protein domain or peptide to generate a molecule with unique structure–function properties and significant therapeutic potential. The gamma immunoglobulin (IgG) isotype is often used as the basis for generating Fc-fusion proteins because of favorable characteristics such as recruitment of effector function and increased plasma half-life. Given the range of proteins that can be used as fusion partners, Fc-fusion proteins have numerous biological and pharmaceutical applications, which has launched Fc-fusion proteins into the forefront of drug development. The topic of Fc-fusion proteins for therapeutic applications is the focus of a newly published book (3).
CD4-Fc represents a classical receptor-ECD–based Fc-fusion protein, in which the protein of interest is fused at the amino terminus (N-terminus) of Fc, replacing the Fab portion of the MAb (Figure 1). The resulting fusion molecule is homodimeric, containing two copies of the ligand-binding domain polypeptide, and it retains IgG Fc effector functions. Typically, the fusion product is expressed using a mammalian-cell system such as Chinese hamster ovary (CHO cells), so that both the protein of interest and the Fc are glycosylated (4). Glycosylation sites in the receptor domains can be N-linked and/or O-linked, whereas those in CH2 of the Fc domain are only N-linked. Because binding to Staphylococcus aureus protein A is retained, purification of the Fc-fusion protein can be achieved using a downstream process that includes protein A capture.
Bioactive moieties can be configured in different ways for Fc-fusion proteins (Figure 2). Typical fusion partners include the ECD of a cell-surface receptor that is fused in frame to the Fc domain of IgG (5). Traps are Fc fusions in which multiple ligand-binding domains from different receptors are fused to the Fc domain. Cytokines (6), enzymes, and growth factors also have been converted to Fc fusions. Monomeric Fc fusions have been described in which only one binding moiety is fused to Fc in a heterodimer (9). Peptides can be fused either at the N-terminus or carboxyl- terminus (C-terminal) of the Fc (as in peptibodies).
Application of Fc-fusion proteins as versatile research tools has been reviewed (7), as have other applications of this class of engineered molecules (5, 8–10). Here we focus on the design and manufacturing of therapeutid Fc-fusion proteins. Since 1998, when the first Fc-fusion protein (Enbrel) was approved by the US Food and Drug Administration (FDA), nine Fc-fusion protein drugs have been approved for market in the United States (detailed in Part 2). Other Fc-fusion proteins are in clinical development (also detailed in Part 2), with still more in preclinical study. We focus on Fc-fusion proteins that have received or will soon receive market authorization, those in clinical testing, and Fc derivatives representing new scaffolds.
Half-Life Extension of Therapeutic Proteins
As mentioned above, the potential biotherapeutic value of many proteins is ultimately limited by rapid in vivo clearance upon intravenous injection, which necessitates multiple weekly administrations. Clearance of therapeutic proteins occurs as a result of metabolism and excretion. Metabolic removal of proteins depends on cellular uptake through receptor-mediated endocytosis or pinocytosis. Removal by excretion depends on a molecule’s hydrodynamic radius, with proteins <60-kDa being rapidly cleared.
Three primary strategies have been used to limit clearance by metabolism and excretion, ultimately improving serum stability of therapeutic proteins: genetic fusion to immunoglobulin Fc, genetic fusion to human serum albumin (11), and chemical conjugation to a hydrophilic polymer such as PEG (12). Fusion to immunoglobulin Fc and albumin extends protein half-life by exploiting the FcRn recycling mechanism (10). Both albumin and IgG are recycled through FcRn (Figure 3), a receptor expressed on vascular endothelial cells. In those cells, IgG and albumin are taken up into endosomes by pinocytosis. When endosome pH is lowered, those molecules associate with membrane- bound FcRn and are transported back to the cell membrane, where they dissociate (depending on pH) from the receptor. Thus, both IgG and albumin are protected from proteolytic degradation and recycled.
Chemical conjugation with PEG increases the hydrodynamic radius of a protein, thereby reducing kidney elimination. PEGylation of therapeutic proteins typically involves amino groups of lysine side chains, resulting in molecular heterogeneity of the purified, conjugated product. PEGylation through thiol groups allows for more limited reactions (13, 14).
Molecular Design
As stated previously, Fc-fusion proteins are MAb derivatives. All MAbs contain Fc domains and have extended in vivo half lives, but several aspects of molecular design distinguish the Fc-fusion protein class. Because such proteins are derived from human receptor and immunoglobulin domains, they are fully human. They offer design flexibility: Typically bivalent homodimers, they also can be monovalent or multivalent. And their ligand affinity can be very high, particularly in the case of cytokine traps, which are homodimers with multiligand-binding domains derived from different receptor chains. The ligand-binding domains combine functionally to form a single high- affinity binding site, thus “trapping” a ligand between those two chains. For example, aflibercept (Eylea) binds monovalently to its ligand VEGF with a Kd of 0.5 pM (15).
To engineer an Fc fusion protein, truncation of the receptor domains is often desirable to reduce the size and structural complexity of the resulting fusion construct — particularly for membrane glycoproteins with large multidomain structures. For Type I transmembrane proteins such as CD4 or TNF receptor, ligand binding is localized within the ECD. All or part of that domain is used to construct the fusion protein, which ensures that binding specificity and/or affinity are maintained. Construction of the fusion molecule is performed at the DNA level, creating a cDNA that encodes the fusion polypeptide.
The C-terminus of the protein of interest is often fused to the N-terminus of the Fc portion of human IgG, either directly or with a spacer peptide (16), to create a junction. Fusion of the Ig Fc domain can be to either the N- or C-terminus of a binding protein, depending on which end can be tethered without impeding correct folding or activity (17). The biologically active domain(s) of a protein of interest replaces the Fab
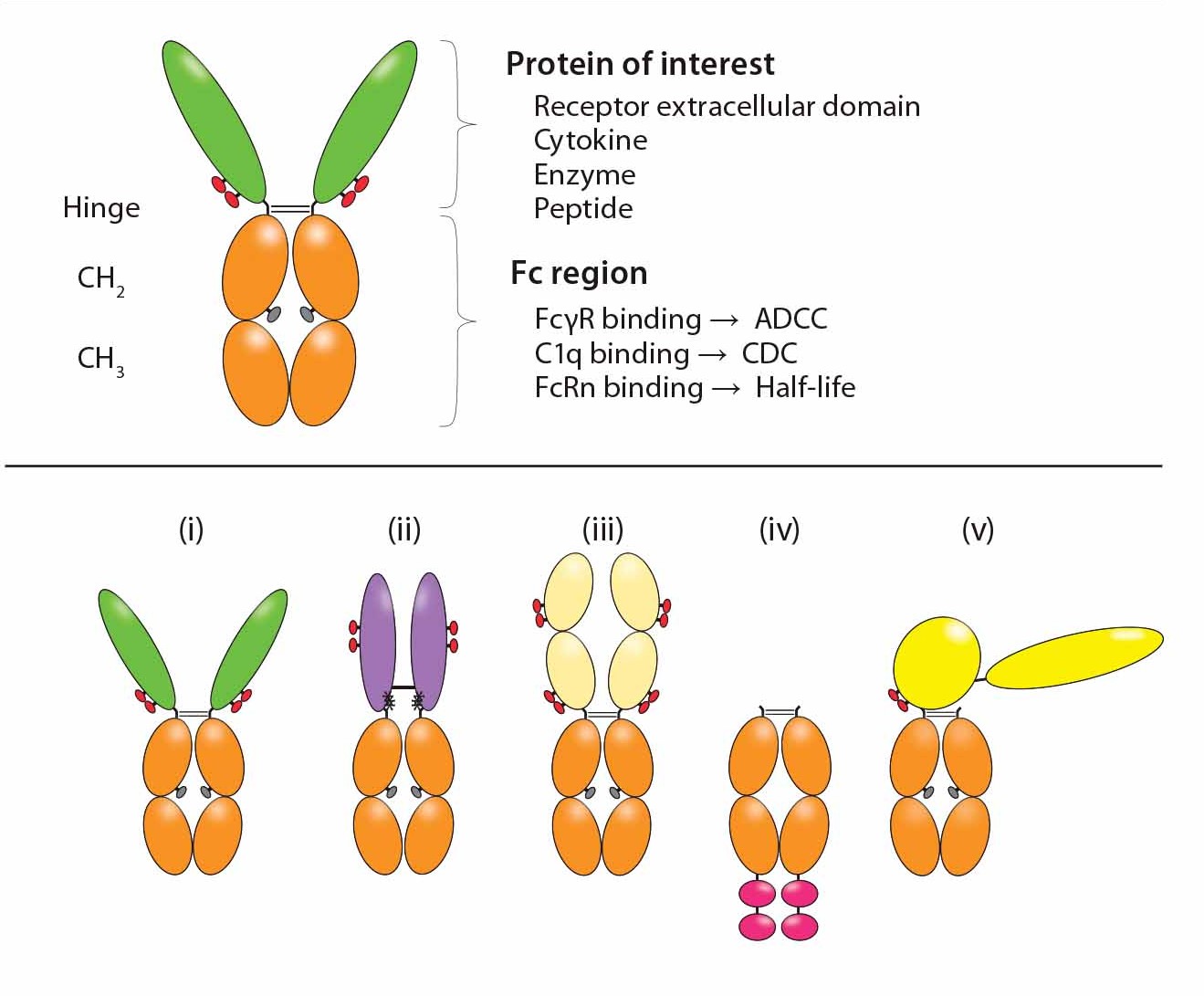
Figure 2: Functional diversity of Fc-fusion proteins; the ligand binding domains can be derived from a receptor ECD, cytokine, enzyme or peptide. The IgG1 Fc region is capable of FcγR and C1q binding, potentially enabling the fusion protein to initiate ADCC, CDC. FcRn binding to the Fc region is responsible for serum half-life extension of the fusion molecule. Reproduced with permission (3).
region — made up of the variable heavy (VH), variable light (VL), and constant heavy 1 (CH1) domains — and is fused with the hinge forming the connection to the Fc region, which is made up of the CH2 and CH3 domains of human IgG1 (Figure 1). The hinge contains disulfide bridges that covalently link heavy chains together, and it differs by IgG subtype in length and in the number of disulfide bonds (18). Note that because fusion proteins are designed to juxtapose protein domains that do not naturally associate, interaction between domains can create physical instability (19). All therapeutic Fc-fusion proteins approved to date are subtype IgG1, and all are capable of binding to Fc receptors.
In the Fc-fusion protein design of CD4 Fc, the site of fusion is located at Asp216 of IgG H chain, which is immediately downstream of the cysteine residue that forms the IgG H–L interchain disulfide bond between the CH1 and CL domains. The hinge serves as a flexible spacer between the two parts of the fusion protein, allowing each part of the molecule to function independently. This fusion strategy maintains structural integrity of the IgG hinge such that, as with MAbs, Fc-fusion proteins can be susceptible to enzymatic cleavage by papain (20).
If a peptide linker is used, the sequence and length must be optimal because it can significantly influence stability of the fusion protein. Zhang et al. outline factors to consider when designing linkers, including methods of design and optimization for fusion- protein construction (21). Alternatively, an enzymatic cleavage site can be engineered into the junction of the fusion protein. Although it is common in Fc-fusion proteins used for research, no such proteins designed as drugs contain an engineered cleavage site. A natural mammalian N-terminal secretory signal sequence is generally used to ensure efficient secretion of the fusion protein from cells during production (22).
Production of Fc-Fusion Proteins Mammalian Expression Systems:
Structurally complex Fc-fusion glycoproteins are mostly produced by mammalian expression systems. They require complex posttranslational modifications — e.g., glycosylation, γ-carboxylation, sulfation, and intra- and interchain disulfide-bonded domains — to maintain their structure and function. All commercial Fc-fusion proteins except for one (romiplostim) are manufactured using mammalian-cell–based processes. CHO cells are the most popular cell line for expression of Fc-fusion proteins because they adapt well to serum-free culture, offer high-titer expression, and facilitate scale-up. Most commercially manufactured Fc-fusion proteins are expressed in genetically stable CHO cell lines (23) using fed-batch cell culture processes. Perfusion production also has been described (24). One potential concern with CHO cells is that their glycosylation patterns can differ from those found in human glycoproteins purified from natural sources. CHO cells add α2,3-linked terminal sialic acid to N-glycans using α2,3-sialyltransferase (ST3), but they lack the ability to add sialic acid to the sixth position (α2,6) of N-linked glycans, which is a hallmark feature of human glycosylated proteins. To overcome those problems, some CHO cell lines have been engineered to express human glycosyltransferases (25).
One commercial Fc-fusion protein (Alprolix) is made using human embryonic kidney 293 (HEK293) cells due to a particular posttranslational processing requirement (γ-carboxylation) for activity of the fusion protein that is unique to HEK293 cells (26). The same cell line also has been used to produce β-secretase Fc (27). Other cell lines such as mouse myeloma (NS0 or SP2/0) cells, monkey kidney (VERO) cells, and the PER.C6 human retinal cell line (28) also have been used but not yet for commercial Fc fusion proteins.
Microbial Expression Systems:
Smaller bioactive peptides fused to Fc (e.g., peptibodies) that do not require posttranslational modifications have been produced using microbial systems. Romiplostim (AMG 531) is expressed by Escherichia coli. A facility for rapid and economical scale-up for protein production makes E. coli suitable for commercial manufacturing (29). Targeted codon optimization has been shown to improve microbial
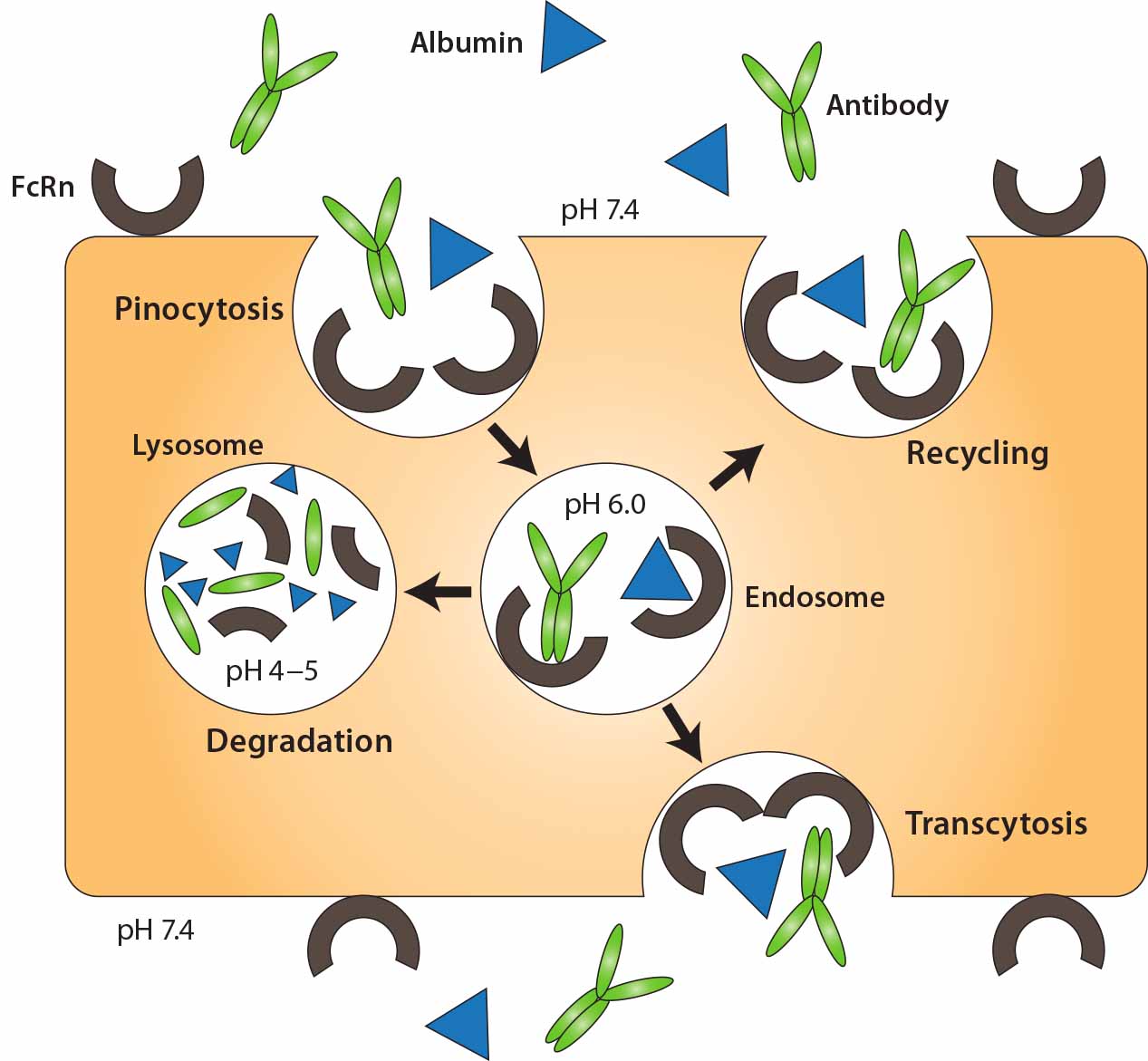
Figure 3: FcRn-mediated recycling of IgG and albumin in vascular endothelial cells; IgG and albumin are internalized into vascular endothelial cells through pinocytosis. The pH of the endosome is 6.0, facilitating association with membrane-bound FcRn. The contents of endosomes can be processed in one of two ways: either recycling back to the apical cell membrane or transcytosis from the apical to the basolateral side. In the case of saturated receptors, excess IgG and albumin are degraded by lysosomes. Top, apical side; bottom, basolateral side. Reproduced with permission from (11).
expression of Fc fusions (30). Although microbial systems have been applied successfully to produce peptibodies, the inability of microbes to posttranslationally modify and thus retain Fc-fusion protein bioactivity has prevented their more widespread use.
Purification of Fc-Fusion Proteins
In manufacturing processes based on mammalian cell culture, Fc-fusion proteins can be purified using a platform downstream process similar to that used for MAbs. Purification of Fc fusion proteins from mammalian cell expression systems typically involves harvest by centrifugation and/or filtration to yield a clarified supernatant. The fusion protein is then substantially enriched with protein A chromatography before polishing with additional chromatographic methods. Typically, protein A is preferred over protein G (which involves stronger binding to Fc and thus requires lower elution pH and offers a lower step yield) for both MAb and Fc-fusion purification. A consideration in scaling the protein A capture step is that Fc-fusion proteins bind to protein A with lower stoichiometry than do MAbs (31), which translates into lower binding capacity of columns.
Each subsequent purification step (32, 33) is chosen based on a given fusion protein’s structural characteristics — e.g., isoelectric point and hydrophobicity profile — to remove additional protein and nonprotein impurities. Low-pH conditions and nanofiltration are incorporated as virus inactivation steps, and ultrafiltration/diafiltration (UF/DF) is used for final adjustment of product concentration and introduction of a formulation buffer.
Challenges in purification of Fc fusion proteins using protein A chromatography include optimizing the elution buffer to maintain biological activity and minimizing aggregation of the protein, using an appropriate nanofiltration technique for virus removal, and identifying polishing techniques that enable both good purity and yield. Fc-fusion proteins often are more extensively glycosylated than are MAbs. The latter typically contain a single glycosylation site in IgG Fc (Asn297 in the CH2 domain). In Fc-fusion proteins, receptor domains generally contain one or more glycosylation sites (both N- and O-linked). Oligosaccharide structures can be more varied (complex and high mannose; bi-, tri-, and tetraantennary) in their receptor domains than IgG Fc (complex, bianntennary) and can contain more sialic acid residues. The latter quality can shift the pI of Fc-fusion proteins into an acidic pH range and impart significantly more charge heterogeneity on them than MAbs have (34).
Fc-Fusion Proteins as BioPharmaceutical Compounds
Currently nine Fc-fusion proteins are FDA-approved and marketed in the United States as biopharmaceutical compounds. Other compounds are in late-stage clinical development. For several clinical indications (e.g., autoimmune conditions such as rheumatoid arthritis), Fc-fusion proteins compete with MAbs designed to bind with similar molecular targets. Eight of the nine currently FDA- approved Fc-fusion proteins are homodimers, and all can be categorized by ligand specificity and valency. Next month, we will conclude with details on these products and the categories into which they are grouped.
Acknowledgments
The authors acknowledge Laura Shih for her assistance with graphics and have no conflicts of interest in publishing this article herein.
References
1 Kontermann RE. Strategies for Extended Serum Half-Life of Protein Therapeutics. Curr. Opin. Biotechnol. 22, 2011: 868–876.
2 Capon DJ, et al. Designing CD4 Immunoadhesins for AIDS Therapy. Nature 337, 1989: 525–531.
3 Therapeutic Fc Fusion Proteins. Chamow SM, et al., Eds. Wiley-Blackwell: Hoboken, NJ, 2014.
4 Sheeley DM, et al. Characterization of Monoclonal Antibody Glycosylation: Comparison of Expression Systems and Identification of Terminal Alpha-Linked Galactose. Analyt. Biochem. 247, 1997: 102–110.
5 Huang C. Receptor-Fc Fusion Therapeutics, Traps, and MIMETIBODY Technology. Curr. Opin Biotechnol. 20, 2009: 692–699.
6 Muller D. Antibody-Cytokine Fusion Proteins for Cancer Immunotherapy: An Update on Recent Developments. BioDrugs 28, 2014: 123–131.
7 Flanagan ML, et al. Soluble Fc Fusion Proteins for Biomedical Research. Monoclonal Antibodies: Methods and Protocols. Albitar M, Ed. Humana Press: New York, NY, 2007; 33–52.
8 Beck A, Reichert JM. Therapeutic Fc-Fusion Proteins and Peptides As Successful Alternatives to Antibodies. MAbs 3, 2011: 415–416.
9 Czajkowsky DM, et al. Fc-Fusion Proteins: New Developments and Future Perspectives. EMBO Mol Med 4, 2012: 1015–1028.
10 Rath T, et al. Fc-Fusion Proteins and FcRn: Structural Insights for Longer-Lasting and More Effective Therapeutics. Crit. Rev. Biotechnol. 2013: 1–20.
11 Weimer T, et al. Recombinant Albumin Fusion Proteins. Fusion Protein Technologies for Biopharmaceuticals. Schmidt S, Ed. John Wiley and Sons: Hoboken, NJ, 2013; 163–178.
12 Fishburn C. The Pharmacology of PEGylation: Balancing PD with PK to Generate Novel Therapeutics. J. Pharmaceut. Sci. 2007: 1–17.
13 Junutula JR., et al. Site-Specific Conjugation of a Cytotoxic Drug to an Antibody Improves the Therapeutic Index. Nat. Biotechnol. 26, 2008: 925–932.
14 Wu P, et al. Site-Specific Chemical Modification of Recombinant Proteins Produced in Mammalian Cells By Using the Genetically Encoded Aldehyde Tag. Proc. Nat. Acad. Sci. USA 106, 2009: 3000–3005.
15 Privalov PL. Stability of Proteins: Small Globular Proteins. Adv. Prot. Chem. 33, 1979: 167–241.
16 Crasto CJ, Feng JA. LINKER: A Program to Generate Linker Sequences for Fusion Proteins. Prot. Eng. 13, 2000: 309–312.
17 Cox G, et al. Enhanced Circulating Half-Life and Heatopoietic Properties of a Human Granulocyte Colony-Stimulating Factor/Immunoglobulin Fusion Protein. Exper. Hematol. 32, 2004: 441–449.
18 Saphire EO, et al. Contrasting IgG Structures Reveal Extreme Asymmetry and Flexibility. J. Mol. Biol. 319, 2002: 9–18.
19 Fast JL, et al. Physical Instability of a Therapeutic Fc Fusion Protein: Domain Contributions to Conformational and Colloidal Stability. Biochem. 48, 2009: 11724–11736.
20 Chamow SM, et al. Enzymatic Cleavage of a CD4 Immunoadhesin Generates Crystallizable Biologically Active Fd-Like Fragments. Biochem. 29, 1990: 9885–9891.
21 Zhang J, et al. Design and Optimization of a Linker for Fusion Protein Construction. Progr. Nat. Sci. 19, 2009: 1197–1200.
22 Kober L, et al. Optimized Signal Peptides for the Development of HighExpressing CHO Cell Lines. Biotechnol. Bioeng. 110, 2013: 1164–1173.
23 Zhang RY, Shen WD. Monoclonal Antibody Expression in Mammalian Cells. Meth. Mol. Biol. 907, 2012: 341–358.
24 Muda M, et al. Therapeutic Assessment of SEED: A New Engineered Antibody Platform Designed to Generate Mono- and Bispecific Antibodies. PEDS 24, 2011: 447–454.
25 El Mai N, et al. Engineering a HumanLike Glycosylation to Produce Therapeutic Glycoproteins Based on 6-Linked Sialylation in CHO Cells. Meth. Mol. Biol. 988, 2013: 19–29.
26 Powell JS, et al. Safety and Prolonged Activity of Recombinant Factor VIII Fc Fusion Protein in Hemophilia A Patients. Blood 119, 2012: 3031–3037.
27 Lullau E, et al. Comparison of Batch and Perfusion Culture in Combination with Pilot-Scale Expanded Bed Purification for the Production of Soluble Recombinant BetaSecretase. Biotechnol. Prog. 19, 2003: 37–44.
28 Ross D, et al. Production and Characterization of a Novel Human Recombinant Alpha-1-Antitrypsin in PER.C6 Cells. J. Biotechnol. 162, 2012: 262–273.
29 Shimamoto G, et al. Peptibodies: A Flexible Alternative Format to Antibodies. MAbs 4, 2012: 586–591.
30 Hutterer KM, et al. Targeted Codon Optimization Improves Translational Fidelity for an Fc Fusion Protein. Biotechnol. Bioeng. 109, 2012: 2770–2777.
31 Ghose S, et al. Binding Capacity Differences for Antibodies and Fc-Fusion Proteins on Protein A Chromatographic Materials. Biotechnol. Bioeng. 96, 2007: 768–769.
32 Lullan E, et al. Comparison of Batch and Perfusion Culture in Combination with Pilot-Scale Expanded-Bed Purification for the Production of Soluble Recombinant BetaSecretase. Biotechnol. Progr. 19, 2003: 37–44.
33 Ross D, et al. Production and Characterization of a Novel Human Recombnant Alpha-1 Antitrypsin in Per.C6 Cells. J. Biotechnol. 162, 2012: 262–273.
34 Beck A, et al. Trends in Glycosylation, Glycoanalysis and Glycoengineering of Therapeutic Antibodies and Fc-Fusion Proteins. Curr. Pharmaceut. Biotechnol. 9, 2008: 482–501.
Angela L. Linderholm is a consultant, and corresponding author Steven M. Chamow, PhD, is president and principal consultant of Chamow & Associates, Inc., 747 Laurelwood Drive, San Mateo, CA 94403; 1-650-345- 1878; steve@chamowassociates.com; www.chamowassociates.com
Thank you for this interesting article. I just wanted to let you and the readers know that there is an alternative to Protein A for purification of Fc-fusion proteins. We, at Thermo Fisher Scientific, have developed an affinity resin targeting the CH3 domain of a human antibody, offering an affinity resin that is able to elute at near neutral pH conditions making it extremely suitable for Fc-fusion proteins. More on this product, called CaptureSelect™ FcXL, can be found on our thermofisher.com website.