Monoclonal antibodies (MAbs) are increasingly formulated at concentrations >100 mg/mL as a means to deliver a high dose in a low volume (1,2). Such high-concentration solutions are commonly opalescent (3,4), an undesirable characteristic of biopharmaceutical products for several reasons. Although it may be only aesthetic, opalescent products are not considered pharmaceutically “elegant.” Of more serious concern, opalescence may be a precursor to aggregation and indicate a propensity toward decreased product stability or quality.
The term opalescent refers to a uniform haze or turbidity in a solution as opposed to the presence of distinguishable particles. Filtering opalescent solutions generally does not decrease their turbidity. The main challenge in studying such protein formulations stems from their forming only at high concentrations (5). Most biophysical characterization techniques require dilution to PRODUCT FOCUS: HIGH-CONCENTRATION ANTIBODIES AND OTHER PROTEINS
PROCESS FOCUS: MANUFACTURING
WHO SHOULD READ: FORMULATIONS, PRODUCT DEVELOPMENT, QA/QC, AND ANALYTICAL PERSONNEL
KEYWORDS: IGG, FTIR, CALORIMETRY, DESIGN OF EXPERIMENTS, STABILITY, TURBIDITY, VISCOSITY, LIGHT SCATTERING
LEVEL: INTERMEDIATE

ONLINE SUPPORTING MATERIAL
Seven more figures providing data to support the results and conclusions drawn in this article can be found online at www.bioprocessintl.com/bpiextra.
Previous work on high-concentration IgG MAb solutions has shown that they commonly form weak, reversible self-associations (6,7) leading to either increased opalescence or viscosity (8,9). However, high-concentration MAbs do not always form oligomers. For example, Sukumar et al. determined that opalescence at high concentration for an IgG1 antibody was solely due to Raleigh scatter (3). The aim of that work was to answer the following questions: Does opalescence lead to decreased product stability or irreversible aggregate formation? What interactions contribute to opalescence? Can it be controlled by careful formulation design?
“MAb X” is a model IgG monoclonal antibody that forms opalescent solutions at high concentrations. We present here two studies on opalescent MAb X solutions to address the above questions. In the first study, solutions were monitored over one month to assess changes in solution turbidity, viscosity, and percent antibody monomer or hydrodynamic radius over time. In the second, we carried out a design of experiment (DoE) to examine the effects of buffer identity, buffer concentration, and NaCl concentration on opalescence. We analyzed select samples with differing turbidities by intrinsic fluorescence, isothermal titration calorimetry (ITC), and Fourier-transform infrared (FTIR) spectroscopy to determine whether secondary or tertiary structural changes could be correlated with opalescence.
Materials and Methods
Concentration and Dialysis of MAb X: We concentrated MAb X from 50 mg/mL to 200 mg/mL in its formulation buffer using a Millipore laboratory-scale tangential-flow filtration (TFF) system with three 30-kDa Pellicon XL filters attached (www.millipore.com). Feed pressure was maintained at 20 psi, retentate pressure at 10 psi. Filters were equilibrated with buffer before addition of the MAb X solution.
We removed aliquots of MAb X from the reservoir periodically during filtration to measure the protein concentration by A280. We dialized 200 mg/mL MAb X into the buffers of interest in prehydrated Pierce Slide-A-Lyzer 3- to 12-mL or 0.5- to 3-mL dialysis cassettes, MWCO 7 kDa, from Thermo Fisher Scientific Inc. (www.piercenet.com). Filled cassettes were then immersed in beakers filled with buffer and placed on stir plates at 2–8 °C. The dialysis cassettes were equilibrated twice for two-hour periods and overnight. We changed the dialysis buffer between equilibration periods, using a minimum total dialysis buffer of 200× the sample volume. All samples were stored at 2–8 °C.
We measured MAb X solution turbidity with a 2100N nephelometer from Hach Company (www.hach.com) with an adapter for small sample volumes. The instrument was calibrated with a Hach Formazin StablCal calibration set over a range of 0.1–4,000 NTU before each use.
Sample aliquots of 3 mL were added to Fisher brand 13-mm glass test tubes. They were coated with a drop of silicon oil and placed into the small-volume adapter. Turbidity was recorded when the reading steadied. We took measurements at 25 °C and 5 °C. For the 5 °C measurements, we kept the instrument under a nitrogen purge to prevent condensation on the sample tubes. Samples were equilibrated at the measurement temperature for a minimum of two hours before measurement.
We measured viscosities with a DV-III Ultra LV rheometer from Brookfield Engineering Laboratories, Inc. (www.brookfieldengineering.com) at 25 °C and 5 °C using a CPE-40 and CP-52 spindle, respectively. Samples were equilibrated for two hours at the measurement temperature before a 0.5-mL sample was placed in the reservoir. The viscosity of each sample was measured at the same five shear rates ranging 75–450 s–1, and the sample was held at each shear rate for one minute. Viscosities varied a maximum of 5% from the minimum to maximum shear rate and thus were averaged to obtain the reported values.
We performed dynamic light scattering (DLS) on 10- and 150-mg/mL samples at 5 °C and 25 °C using a DynaPro plate reader with Dynamics software (version 6.9.2.11) from Wyatt Technology Corporation (www.wyatt.com). For the 5 °C measurements, we purged the instrument with 60 psi nitrogen to prevent condensation. We prepared samples of 10-mg/mL MAb X by dilution of 150-mg/mL MAb X with matching buffer. A 75-L aliquot of each sample was pipetted into a well of a 384-well, low-volume microplate from Corning Life Sciences (www.corning.com).
We used the following settings: laser power 100% (10-mg/mL samples) or 25% (150-mg/mL samples), 10 acquisitions at 10 seconds acquisition time. Buffer properties were assumed to be the same as those of phosphate buffered saline (PBS). We analyzed the data with Dynamics software using a globular protein model, using hydrodynamic radii for the 150-mg/mL samples solely for comparison of results of 150-mg/mL samples across different buffers.
We used a Cary Eclipse fluorescence spectrophotometer from Varian, Inc. (www.varianinc.com) to collect intrinsic fluorescence emission spectra of 0.1 mg/mL MAb X. Excitation was at 292 nm, with emission monitored from 300 to 450 nm at a scan rate of 120 nm/min in a 1-cm path length cuvette. Sensitivity was set high, and both excitation and emission slits were 5 nm wide. We subtracted the corresponding buffer spectrum from each sample spectrum and calculated the center of mass (COM) using the following equation, where λ is the wavelength, and Iλ is the emission intensity at wavelength λ:
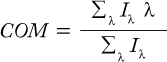
We took Fourier-transform infrared (FTIR) spectroscopy measurements of 150-mg/mL MAb X solutions at 25 °C using a Nicolet 4700 instrument with a liquid cell from Thermo Scientific (www.thermo.com) and analyzed the spectra with its accompanying Omnic software. We used the following settings to collect background, sample spectra, and buffer spectra: autogain, resolution = 4, final format = absorbance, number of scans = 256. Background was collected every 120 minutes. We subtracted buffer spectra from the corresponding protein spectra using manually adjusted smoothing and took the Savinsky-Golay second derivative with 11 points.
We performed isothermal titration calorimetry (ITC) at 25 °C using an iTC200 from MicroCal, part of GE Healthcare (www.microcal.com). The titrant syringe was filled with 40-µL of titrant: either 150 mg/mL MAb X, 50 mM 8-anilinonaphthalene-1-sulfonic acid ammonium salt (ANS), or buffer. The sample cell was filled with either 200 µL of buffer or 150 mg/mL MAb X. The ANS solution was prepared by mass with >97% purity Fluka Biochemika ANS from Sigma (www.sigmaaldrich.com) and buffer from protein dialysis.
Each run consisted of 39 injections added at 0.5 µL/s. The injection volume was 1.0 µL for all injections except the first, which was 0.1 µL. We used the following settings: reference power = 5 µcal/s, initial delay = 60 s, feedback mode/gain = high, filter period = 5 s, stir speed = 1,000 rpm, and injection spacing = 410 s. Control runs included buffer titration into buffer, 10 mM ANS titration into buffer, and buffer titration into 150 mg/mL MAb X. We subtracted the corresponding control runs from each sample run and analyzed the data with Origin software (version 7.0, www.originlab.com). Because fits to the one-site and two-site models resulted in statistically insignificant parameter values for the number of sites (n), the association constant (K), and enthalpy of binding (ΔH), we used the ANS binding curves solely for comparison across different buffers.
We quantified percent monomer by size-exclusion chromatography (SEC) using an HP-1100 system from Agilent Technologies, Inc. (www.agilent.com) with a TSK-GEL G3000SWxI column from Tosoh Bioscience LLC (www.tosohbioscience.com). The mobile phase contained 100 mM NaPO4 with 400 mM NaCl and was set at a flow rate of 1 mL/min. Two individual 100-µL injections were made of each 1-mg/mL sample. The chromatograms were integrated using Empower software from Waters Corporation (www.waters.com) to determine monomer percent. We compared the total area of those chromatograms with that of a reference standard to verify complete sample recovery from the column.
Design of Experiment: We set up response surface central composite design using Design Expert software (version 7.1) from Stat-Ease, Inc. (www.statease.com) to examine three factors: buffer identity, buffer concentration, and NaCl concentration. The numerical factors (buffer concentration and salt concentration) had two levels, and the categorical factor (buffer identity) had three levels, and there were two center points per categorical factor level. Solution pH was held constant at 5.5. Responses measured were turbidity, viscosity, percent antibody monomer, and fluorescence COM.
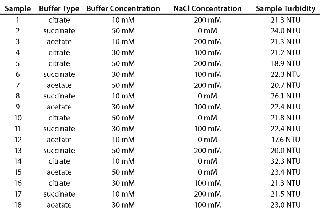
Table 1 shows the DoE design, with turbidity of 150-mg/mL MAb X in each buffer. We dialized a 3-mL aliquot of 200 mg/mL MAb X into each buffer, and the resulting concentration ranged 140–160 mg/mL for the 18 samples. Each sample was diluted to a final concentration of 140 mg/mL with matched buffer. All solution preparations and measurements were performed in the order listed in Table 1.
Table 1: Formulations used for DoE and measured turbidities of 150-mg/mL samples
Results and Discussion
Figure 1 shows the turbidity of MAb X as a function of concentration in pH 7 citrate and pH 6.0 acetate over a range of buffer concentrations at 25 °C. We generated those curves by starting with high-concentration MAb X and diluting with matching buffer. Qualitatively identical results were obtained at 5 °C, with the turbidity shifted to higher values. Several features of MAb X turbidity are evident in Figure 1. Below ~50 mg/mL, turbidity was linear and independent of buffer type, concentration, and possibly pH.
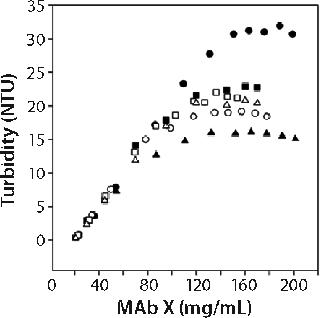
Linear dependence of turbidity on concentration (or Raleigh scattering) is expected for ideal solutions (10). At higher concentrations, the turbidity of MAb X solutions was higher in citrate buffer than in acetate at both 5 °C and 25 °C. Turbidity decreased with increasing citrate concentration while remaining independent of acetate concentration. At ~100 mg/mL, it reached a plateau in all buffers. This phenomenon does not appear to have been previously reported, but the nonlinearity of turbidity with concentration suggests that protein interact
ions were present (10). The plateau value depended on temperature, buffer, and buffer concentration. We systematically examined the roles of buffer identity and concentration in contributing to opalescence through a DoE approach.
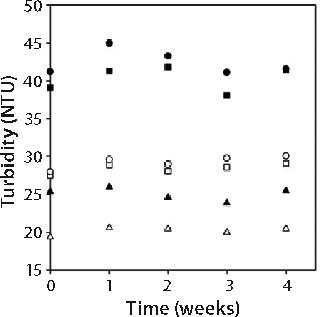
Stability of Opalescent MAb X Solutions: We evaluated stability of high-concentration MAb X formulations with a range of turbidities to determine whether opalescence is indicative of decreased stability. We placed 1-mL samples of 150-mg/mL MAb X in 10 mM citrate at pH 7, 10 mM citrate at pH 6.5, and 20 mM acetate at pH 6.0 in hermetically sealed glass vials and stored at 5 °C and 25 °C for one month. A sample from each storage condition was pulled weekly and evaluated by SEC and DLS as well as for turbidity and viscosity.
Figure 2 shows turbidities of 150-mg/mL MAb X solutions. Turbidity values were independent of storage time and inversely related to storage temperature. Such an observed temperature dependence has been reported for other MAbs (3). As in Figure 1, turbidity was markedly lower for MAb X solutions in acetate buffer than in citrate buffer.
The viscosity of 150-mg/mL MAb X solutions also was independent of storage time. We found no difference in viscosity for protein solutions in the three buffers (data not shown). Viscosity of 150-mg/mL MAb X at 25 °C was 8 ± 1 cP, and at 5 °C it was 30 ± 5 cP.
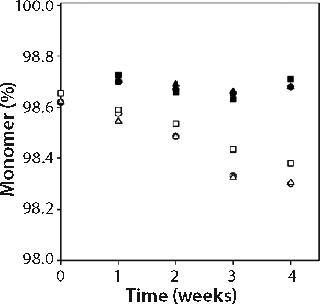
We measured percent monomer by SEC after diluting samples to 1 mg/mL; therefore all aggregates observed were irreversible by dilution. Percent monomer was constant at 99.7 ± 0.1% for samples kept at 5 °C storage, but for samples stored at 25 °C it dropped over the four-week study from 99.7% to 99.3% in all three buffers (see online supporting material, Figure 1). The increase in aggregate from 0.3% to 0.6% over four weeks storage at 25 °C did not, however, correspond to an increase in turbidity, nor was there a larger percentage of aggregate formed in the formulations with higher turbidity (citrate buffer formulations) than in those with lower turbidity (acetate buffer formulations). Combined SEC and turbidity data indicate that irreversible aggregates were not responsible for causing the opalescence of MAb X solutions.
We performed DLS on both 150-mg/mL samples and those diluted to 10 mg/mL with matched buffer. Figure 3 shows the apparent hydrodynamic radius from these measurements. The radii values reported for the 150-mg/mL samples is not the true value because of complications associated with performing DLS on high-concentration samples. We assumed that effects of multiple scattering or high viscosity on the calculated hydrodynamic radii were the same on all three samples because we took our measurements in the three different buffers on samples with the same concentration. Thus we used the reported values solely for comparison among the samples.
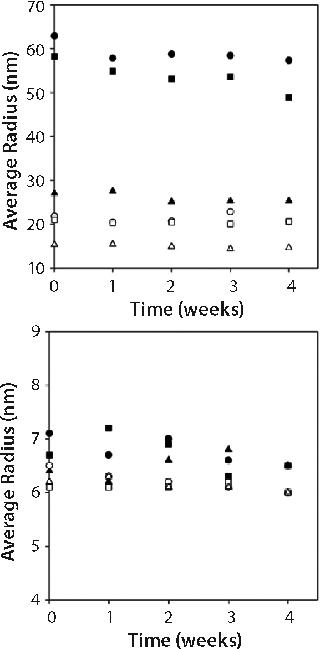
Our attempts to correct the measured hydrodynamic radius values for solution viscosity using measured viscosities resulted in unreasonably small radii values corresponding to molecular weights of only half the monomer’s molecular weight. Therefore, we used the viscosity of phosphate buffered saline (PBS) in the Dynamics software to calculate the radii reported.
Figure 3 suggests that the apparent hydrodynamic radii of 150-mg/mL samples were lower in acetate than in citrate buffer systems. But on dilution to 10 mg/mL, the hydrodynamic radii were the same regardless of buffer type. This is consistent with MAb X solutions having lower turbidity in acetate than in citrate at high concentration and converging to the same turbidity at concentrations
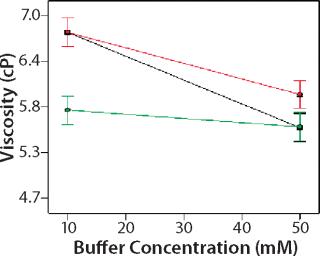
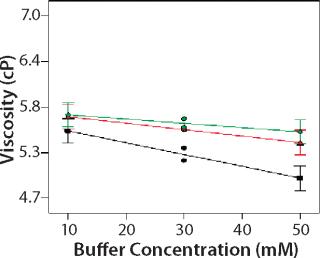
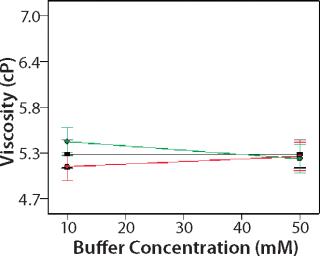
Design of Experiment: We used a DoE to investigate the role of buffer identity, buffer concentration, and salt concentration on MAb X opalescence at pH 5.5. Table 1 lists the 18 formulations examined and their turbidity values at 150 mg/mL. We measured viscosity, percent monomer, and turbidity at 25 °C and analyzed the results with Design Expert software as described above. Viscosity ranged from 5 to 7 cP for all 18 formulations, and the trends correlated well with the turbidity. Viscosity interaction plots can be found online in supporting materials Figures (234). Percent monomer variation among the samples was not statistically significant: All samples had values between 99.7% and 99.8% (data not shown). These results further indicate that soluble aggregates do not correlate with MAb X turbidity.
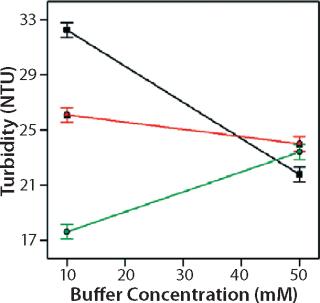
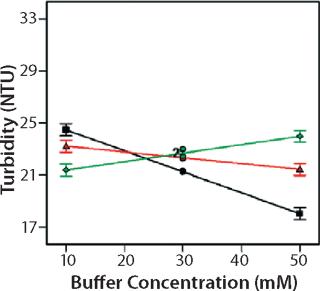
Role of Buffer Concentration, Salt Concentration, and Buffer Identity:456 show interaction plots for turbidity plotted against buffer concentration for 150-mg/mL MAb X in citrate, succinate, and acetate with 0 mM NaCl (Figure 4), 100 mM NaCl (Figure 5), and 200 mM NaCl (Figure 6) at 25 °C. The plots for 5 °C (data not shown) showed the same trends but at higher turbidity values. Data from Figures 456 indicate that the dependence of MAb X turbidity on buffer and salt concentrations was specific to each buffer.
on:none;background-color: #f7f7f7;color:#000000;” >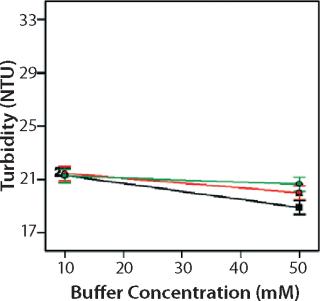
In citrate, increased buffer concentration and salt concentration both independently lowered turbidity of MAb X. The same trend was apparent in succinate, but it was much weaker and near the standard measurement error. In acetate, however, increased turbidity correlated with increasing acetate concentration and NaCl concentration at low ionic strengths. Previous work has shown that MAbs can exhibit either an increase or decrease in turbidity with increasing ionic strength (4,7,9,11). We found the effect of changing buffer concentration to be most evident in all three buffers at low salt concentration. The opposite was also true: Effects of changing salt concentration were most pronounced at low buffer concentrations. These findings indicate that overall ionic strength was a driving factor in opalescence, not simply buffer or salt concentrations alone.
Ionic strength may have affected turbidity through several mechanisms. The first of these is shielding of electrostatic interactions between protein molecules. If those interactions were the only factor involved, then the dependence of turbidity upon ionic strength should be equivalent in all three buffers. But the behavior of MAb X in acetate was qualitatively different than in citrate and succinate. Additionally, the temperature dependence of turbidity indicated that electrostatic interactions alone did not govern turbidity (they are not highly temperature dependent). Solution ionic strength and temperature may also have subtly altered protein conformation, thereby affecting hydrophobic interactions and self association.
As Figures (456) show, buffer identity also contributed to MAb X opalescence. However, buffer identity was important only in determining opalescence at low ionic strength. As salt concentration increased, differences among the three buffers diminished to zero. At low ionic strength, the buffers in order of decreasing turbidity were citrate > succinate > acetate, which does not follow their calculated ionic strengths at pH 5.5. It does agree with the DLS results in Figure 3 showing the hydrodynamic radius of 150-mg/mL MAb X to be lower in acetate than citrate.
Kameoka et al. found a similar buffer-specific dependence for aggregation of a humanized IgG MAb (12). It had lower aggregation propensity in acetate than in citrate, which was determined to be due to interactions of the buffer species with hydrophobic regions of the Fc domain of the IgG. Because MAb X is also an IgG, it is possible that acetate could interact with hydrophobic regions of MAb X, preventing self-association. Such interaction could explain the lower turbidity and different dependence on ionic strength of MAb X in acetate buffer.
Role of MAb X Structure: We used biophysical characterization techniques to determine whether structural differences in MAb X could be detected for solutions of differing opalescence. Intrinsic tryptophan fluorescence was used to probe the tertiary structure of the 18 DoE samples. But because the samples had to be diluted to 1 mg/mL for measurement, conformational changes due to reversible associations at 150 mg/mL would not be detected in the spectra. These data may determine only whether the formulation induced a tertiary structure change in the monomer, which could in turn increase associations and turbidity at higher protein concentrations. All 18 samples had overlaying fluorescence emission spectra (data not shown). The COM ranged 332–335 nm, which indicated partial exposure of tryptophan residues to the solvent environment. COM variation among samples, however, was not statistically significant, indicating that opalescence did not correlate with a change in tryptophan exposure.
As an alternative tertiary and quaternary structure probe to fluorescence, we used ITC to conduct two sets of experiments. In the first set, we titrated 50 mM ANS (a molecule that binds to hydrophobic regions of proteins) into 150-mg/mL MAb X. Enthalpograms for ANS binding to MAb X in two formulations of differing turbidities (one acetate and one citrate) were the same within experimental error (see online supporting material, Figure 5), which suggests that the accessible hydrophobic areas of MAb X were similar in the citrate and acetate-buffered samples.
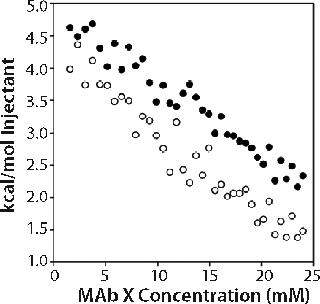
In the second set of experiments, we titrated 150-mg/mL MAb X into matched buffer. The measured heat evolved included both heat of protein dilution and heat of dissociation from oligomers present. We expected comparison of the enthalpograms for dilutions in pH 6.0 20 mM acetate and pH 7.0 10 mM citrate to reveal whether differences in the levels of associated species existed in the two buffer systems. Data shown in the online supporting material (Figure 6) show a significant but equivalent heat of dilution in both buffers. The enthalpograms did not fit a dimer dissociation model, and we did not attempt to fit the data to more complicated dissociation models.
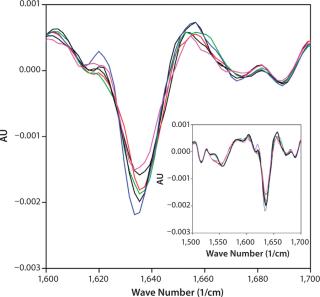
We selected FTIR for secondary structural analysis because of its compatibility with high-concentration solutions. Spectra were measured for each of the 18 DoE samples to determine whether secondary structural differences could be detected among samples of differing turbidity. All 18 spectra nearly overlaid (see online supporting material, Figure 7) and indicated the presence of β-turns (peaks at 1,688 and 1,672 cm−1), β-sheets (1,635 cm−1), and extended β–strands (peak at 1,615–1,617 cm−1) regions (13,14). We saw no shifts in peak position for any sample and no trends in peak magnitudes corresponding to turbidity. FTIR spectra showed no correlation between measurable secondary structural changes and MAb X turbidity.
Opalescence and Stability
One-month stability data indicated that the underlying driver of opalescence is a thermodynamic event rather than a kinetic one. Formulations with higher turbidity did not demonstrate higher levels of aggregate formation than those with lower turbidity. DLS data showed that opalescence of MAb X solutions may be due to reversibly associated species that are larger in citrate buffer than in acetate. The ITC dissociation data were consistent with the presence of reversibly associated species, but we detected no difference among solutions with differing turbidities.
None of the biophysical characterization techniques revealed changes in MAb X structure for any of the formulations we examined, so no tertiary or secondary structural changes could be correlated with opalescence. This indicated that either the structural changes are negligible or that only an immeasurably small percentage of the population forms reversible oligomers. Overall, the data showed opalescence to be a complicated thermodynamic phenomenon that is greatly affected by temperature, buffer identity, and ionic strength. There are probably electrostatic and hydrophobic contributions in addition to buffer-mediated interactions at low ionic strengths. Although the interactions dominating opalescence may be protein-dependent, our work demonstrates that information gleaned from a DoE approach to formulation can be used to minimize opalescence by optimizing the buffer and ionic strength of high-concentration antibody formulations.
Supporting Figure 1: >
Supporting Figure 2:
Supporting Figure 3:
Supporting Figure 4:
Supporting Figure 5:
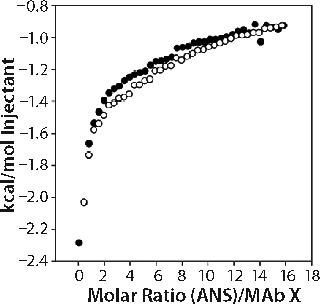
Supporting Figure 6:
Supporting Figure 7:
Author Details
Corresponding author Jennifer M. Woods is principal scientist, and Douglas Nesta is director of biopharm R&D in biopharmaceutical technologies for GlaxoSmithKline R&D, 709 Swedeland Road, King of Prussia, PA 19406; jennifer.m.woods@gsk.com.
REFERENCES
ion of Humanized IgG. J. Biochem. 142:383-391.