 Stability testing is a vital part of product development and is conducted throughout a product’s life cycle (Figure 1). Stability is part of a biotherapeutic’s quality target product profile, and results help analysts understand how critical quality attributes (CQAs) of both drug substances and products are influenced under specific conditions of temperature, relative humidity (RH), light, storage, pH, and other factors. Manufacturers conduct stability tests to determine degradation pathways and establish shelf lives and storage conditions of their products, for example. Tests and bioassays related to purity, identity, potency, quality, and safety are conducted depending on the product type and intended use.
Stability testing is a vital part of product development and is conducted throughout a product’s life cycle (Figure 1). Stability is part of a biotherapeutic’s quality target product profile, and results help analysts understand how critical quality attributes (CQAs) of both drug substances and products are influenced under specific conditions of temperature, relative humidity (RH), light, storage, pH, and other factors. Manufacturers conduct stability tests to determine degradation pathways and establish shelf lives and storage conditions of their products, for example. Tests and bioassays related to purity, identity, potency, quality, and safety are conducted depending on the product type and intended use.
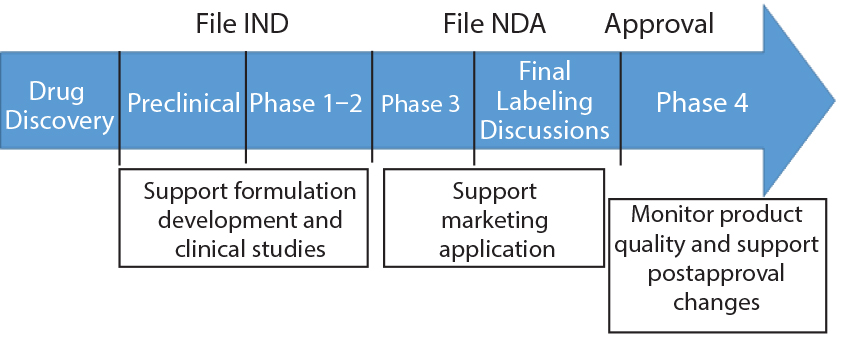
Figure 1: The purpose of stability testing throughout a biologics’s life cycle (22)
This overview presents some current issues with testing the stability of current and next- generation biologics. Although it is impossible to thoroughly cover all aspects of stability testing in this space, many available literary resources and conferences discuss finer details.
Testing in Stages
All stages of a biologic’s life cycle includes stability testing. Experts have divided these tests into six stages, covering development from early stages to late-stage follow-up testing (1, 2):
- Stage 1 (early stage stress and accelerated testing with drug substances)
- Stage 2 (stability on preformulation batches)
- Stage 3 (stress testing on scale-up batches)
- Stage 4 (accelerated and long-term testing for registration purposes)
- Stage 5 (ongoing stability testing
- Stage 6 (follow-up stability testing).
The information that is obtained during testing depends on the stage of development. For example, shelf life is determined during long-term testing, and accelerated testing is conducted to assess product degradation pathways and develop stability-indicating analytical methods. Prototype testing is performed during early stages to characterize a product or formulation. Stability tests also are conducted after reconstitution of a freeze- dried product (3).
In-use stability testing is discussed in a European Medicine Agency note for guidance (4). The described studies mimic how a product is delivered to/used by a patient. In-use testing applies to multidose products (e.g, pen injectors, multidose vials, inhaled biologics), which are at risk of microbial contamination, proliferation, and physicochemical degradation because of the repeated opening and closing of their container-closure systems (4).
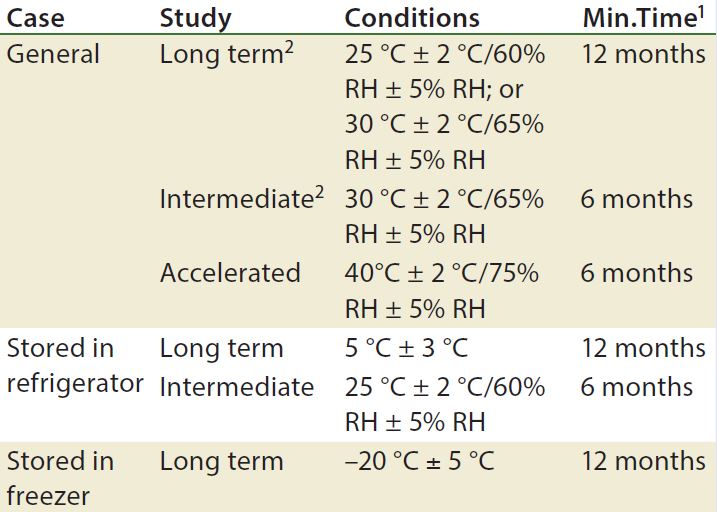
Table 1: Drug products should be evaluated under storage conditions; ICH Q1A guidelines for storage
1 Minimum time period covered by data at submission
2 It is up to an applicant to decide whether long-term stability studies are performed at 25 °C ± 2 °C and 60% RH ± 5% or 30 °C ± 2 °C and 65% RH ± 5% RH
3 If 30 °C ± 2 °C and 65% RH ± 5% RH is the long-term condition, then there is no intermediate condition.
Regulatory Requirements
Stability testing is required by a number of regulatory agencies. The ICH harmonized tripartite guidelines on stability testing are the Q1A–Q1E documents (Q1F was withdrawn in 2006) and ICH Q5C, which is specifically for biologics (3, 5–9). Q1A is the parent guideline for stability testing of all pharmaceuticals. It discusses stress testing, batch selection, testing frequency, storage conditions (Table 1), and other topics. 21 CFR 211.166 also discusses stability testing for finished drug products (10). USP general chapters <1049> and <1046> are informational (not enforceable) guidances that provides general considerations for biopharmaceuticals and cell and gene therapies, respectively (11, 12).
EMA guidelines include CPMP/QWP/609/96: Declaration of Storage Conditions (an annex to stability guidelines and applies to storage condition statements on labeling); CPMP/ QWP/2934/99: In-Use Stability Testing (applies to products in multidose containers); and CPMP/ QWP/159/96: Maximum Shelf Life for Sterile Products After First Opening or Following Reconstitution (which says users are responsible for maintaining the quality of a product that is administered to patients and provides guidance on user-information text) (13–15).
Other organizations such as the World Health Organization and the Association of Southeast Asian Nations have released stability testing guidelines, both for finished products and active pharmaceutical ingredients (APIs), but some apply only to conventional (small-molecule) drug products. Some emerging markets outside the ICH region (e.g., Brazil, China, and South Korea) have written country-specific stability requirements (16).
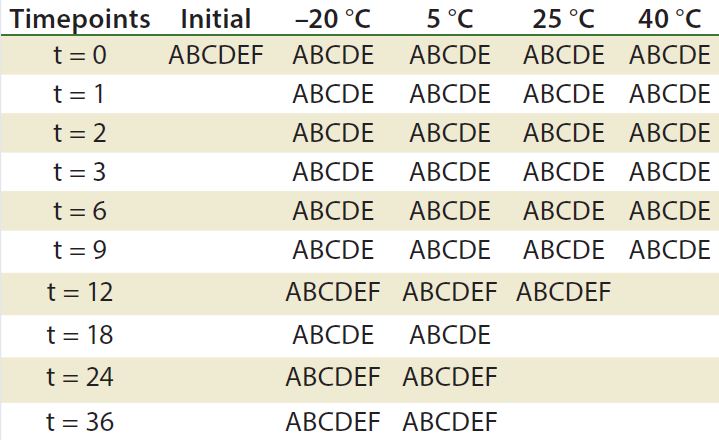
Table 2: Stability ptotocol example for frozen liquid biologic product (17)
A = appearance, pH, UV, particulate (drug product); B = immunoblots and gels such as sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS PAGE) and isoelectric focusing (IEF); C = high-performance liquid chromatography (HPLC) (reverse-phase, ion-exchange, size-exclusion chromatography, hydrophobic interaction chromatography) capillary electrophoresis; D= peptide mapping, mass spectrometry; E = functional assay (enzyme-linked immunosorbent assay, bioassay); F = bioburden (drug substance), sterility, endotoxin (drug product)
Technologies and Protocols
Although stability testing is covered in several guidelines, those documents do not provide details about how to conduct the tests they recommend. Analysts must develop analytical methods and bioassays and then validate them as stability- indicating methods. A stability-indicating protocol is developed according to what is needed to be determined. The protocol dictates when particular methods must be conducted (Table 2). The profile must assure that changes in a product’s identity, purity, and potency will be detected (3). Protein Instability: Stability of a final biopharmaceutical drug product depends highly on the stability of the protein itself. Analysts use a number of bioassays to characterize proteins and verify their function. The EMA has listed common instability issues (18):
- deamidation (hydrolysis of asparagine and glutamine side chain)
- oxidation of methionine (Met), histidine (His), cysteine (Cys), tyrosine (Tyr), and tryptophan (Trp) residues
- aggregation (association of monomers or native multimers covalent or noncovalent)
- glycoproteins (most common instability of glycosylation hydrolysis of sialic acid residues).
Different drug product types have different characteristics that affect their stability. The “MAb” box lists a wide range of physicochemical and biological characteristics that can affect stability of monoclonal antibodies, for example. Other factors influencing stability include the presence of impurities in a formulation and the type of excipients used.
| MAB Characteristics Affecting Stability |
| Physicochemical Variable region: deamidation, oxidation, N-term pyroglutamate (pyro-Glu), glycosylation, glycation Constant region: deamidation, oxidation, acetylation, glycation, glycosylation (e.g., fucosylation, sialylation, galactosylation, mannosylation), C-terminal lysine, disulfide bond shuffling/cleavage, fragmentation/clipping |
| Biological Binding: Affinity, avidity, immunoreactivity or crossreactivity, unintentional reactivity Effector Function: complement interaction, FcRn, FcγR interaction, mannan-binding ligand interaction, mannose receptor interaction Other Properties: Pharmacokinetics, epitope/immunogenicity, modulatory region |
| Reference 1 G. Jimenez, Brake B. ICH Q5C Stability Testing of Biotechnological/Biological Products. EMA Training for ASEAN, 30–31 May 2011; www.ich.org/fileadmin/Public_ Web_Site/Training/ASEAN_Q5C_workshop_May_2011/ SESSION_Ia_ICH_Q5C.pdf. |
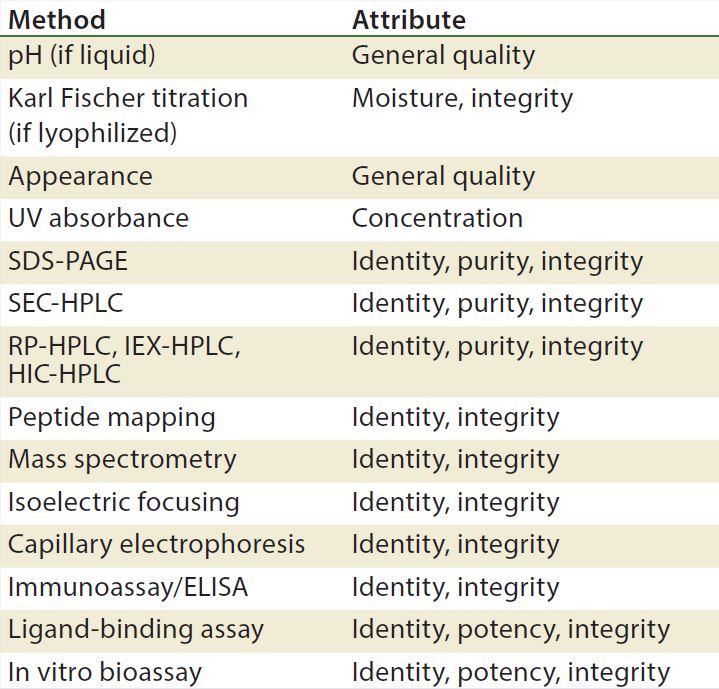
Table 3: Stability test methods for recombinant proteins, antibodies, and blood products (17)
Methods/Technologies: Drug products can undergo physical, chemical, and microbial changes that affect their efficacy. No single bioassay or analytical method can be used to verify a product’s identity, purity, and potency. So a number of orthogonal analytical technologies and bioassays are typically performed, including many chromatography and spectroscopy methods. Tables 3–5 list some methods for particular products. The types of analytics and bioassays used depend on product type, phase of development, and purpose of the test.
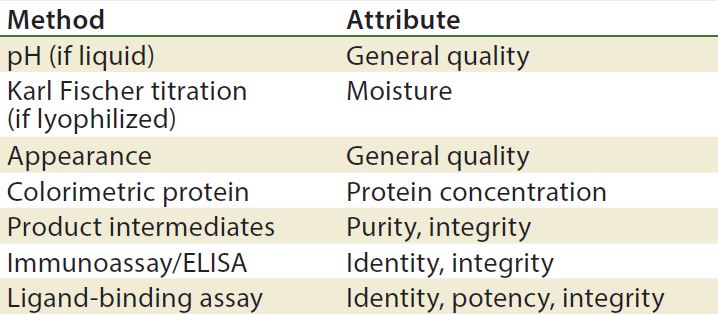
Table 4: Stability test methods, viral vaccine products (17)
For example, stability studies can be a part of a larger comparability assessment. That is used to compare the same drug product after the same manufacturer has made changes or to determine whether a formulation has changed or if a molecule itself has changed during processing (e.g., structure change, deamidation).
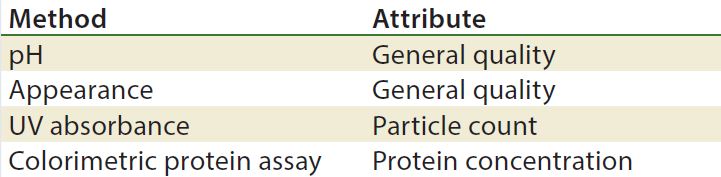
Table 5: Stability test methods, gene therapy viral vectors (17)
Forced degradation studies help companies understand degradation pathways, identify degradation products, develop formulations, and determine a molecule’s instrinsic stability. Analysts conduct these studies by intentionally degrading a molecule to an appropriate extent through stressing conditions of temperature, pH, light, and so on. Currently no guidance or requirement exists about when and how to conduct forced degradation studies.
In accelerated stability testing, products are subjected to high temperatures and relative humidity conditions. Other stress conditions include agitation, light, and moisture. The purpose is to determine the point of product failure by accelerating degradation. Results help companies predict shelf life or compare the relative stability of alternative formulations.
Next-Generation Drug Products
Analysts have been performing stability tests on products such as MAbs and vaccines for some time and — although stability-indicating methods are unique to each product — guidance for such products is available.
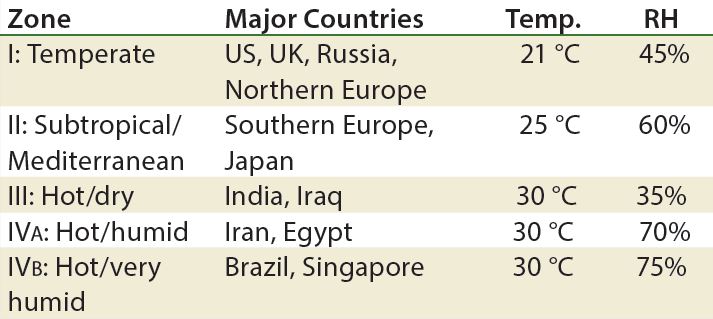
Table 6: Climate zones for drug product “room temperature” storage” (RH = relative humidity)
Next-generation products such as antibody–drug conjugates (ADCs), high- concentration biologics, and cell therapies each have their own challenges. Stability testing of these products can be different from those of well- characterized therapies such as antibodies and some vaccines, especially if marketed globally (Table 6).
ADCs combine the potency of a small-molecule drug (e.g., cytotoxin) with the targetting of a monoclonal antibody. ADC stability testing presents unique challenges (such as with potency and method development) because of the linkerand small-molecule components (as the “Antibody–Drug Conjugates” box explains).
Cell and gene therapies cannot endure storage at room temperature. Noncryopreserved products have much shorter half-lives than other biologics, so analysis on them can be challenging. Cryopreserved products must be thawed, washed, and administered with a cryoprotectant at a clinic. Stability tests are required for IND filing according to 21 CFR 312.23(a)(7)(iii). Cell and gene therapy manufacturing, raw materials, characterization and other aspects are discussed in USP <1046>, and cell and gene products are regulated under the FDA’s good tissue practices final rule.
Testing must include shipping, storage, and in-process holding of cells at all phases of manufacturing. Stability studies are required for cell and virus banks, critical raw materials, reference standards, bulk materials, and final products. In-process tests help analysts determine whether a cell or gene therapy product is stable during cryopreservation, and a comparability test is often conducted on prefrozen and postthaw products.
Stability tests at later phases generate data that support final formulation (19). Purity tests verify that a finished product does not contain extraneous material and residues such as unwanted cellular subsets (21 CFR 600.3(r)). Analytical strategies also include pyrogenicity/endotoxin tests either by rabbit pyrogen method (21 CFR 610.13) or Limulus amebocyte lysate methods (21 CFR 510.9). Potency tests for cell therapies are currently under discussion, and the FDA has issued a draft guidance regarding potency tests for cell and gene therapeutics (20). Final stability testing is used to establish expiration dates and to verify that products are stable under shipping conditions and storage (stress testing).
Combination products are subject to the same general requirements as other drug products. Specific analytical methods and assays depends on the type of drug and device. If the product contains a biologic, then it must undergo the same tests for identity, purity, and potency as an individual biologic product. Studies on medical devices test for stability of matrix materials and ensure that a device does not introduce contaminants. Stability protocols generally dictate long-term studies because of the long shelf lives of most combination products as well as stress testing because of the varying storage conditions for such products.

Figure 2: Particle classification according to size.
Highly Concentrated Formulations: Some patient-administered parenteral biologics are formulated at high concentrations. Such products have a tendency to aggregate and, especially during storage (Figure 2). Aggregation can lead to the formation of particulate matter, which can lead to denaturation, immunogenicity risks, and altered potency (21). Methods to evaluate size and/or concentration include dynamic light scattering, flow imaging, and microscopy. Newer techniques for measuring size and concetration of subvisible particles include resonance-based mass measurement (23). For high-concentration formulations, stability testing is needed throughout development as well as at specific time points during storage. Assessment must be made at the same concentration that will be used for administration. But how well traditional analytical methods work on these types of formulations remains a topic of investigation (24).
|
Stability Testing of Antibody-Drug Conjugates The following is from a discussion with Gentech scientist Colin D. Medley about the stability testing challenges with ADCs. |
| Comparison with MAb Stability Testing: “There are a lot of similarities, including some basic tests such as color, clarity, opalescence, pH, and protein concentration. As with MAbs, aggregation is generally measured by size-exclusion chromatography (a key stability-indicating assay). It’s definitely one parameter that must be controlled. Charge heterogeneity test methods such as capillary isoelectric focusing are used for both MAbs and ADCs. But generally ADCs are more complicated because of more potential sources for charge variation, mostly from the conjugated small molecule.“Where you start to see differences is around the drug. You want to make sure your small molecule (either through process impurities or subsequent degradation) does not deconjugate from the antibody. It then has different pharmacokinetics than a conjugated drug. That is something you really want to minimize with ADCs, particularly with highly potent toxins. Also, you need to check drug distributions and drug-to-antibody ratios. You want to make sure that the drug remains conjugated to the antibody in a sufficient ratio so that it remains efficacious on stability.” |
| Analytical Methods: “Many methods for ADCs can and will be affected by the properties of a small-molecule drug, but it usually does not prevent their use. For protein concentrations, that is a problem we see frequently. The small molecule will have an absorbance in the 280-nm range. So when you are trying to measure protein concentration at the 280-nm signal, you’re not just measuring protein, you are also measuring the small molecule. So your extinction coefficient for the conjugate will be different than it would be for just a naked monoclonal antibody. You’ll usually have to account for that in the method by subtracting out the portion of the absorbance that is related to the small molecule rather than the antibody. And that can be a bit more complicated when doing stability studies if you see degradation in the small molecule.“ |
| Degradation Pathways: “An ADC degrades much like a MAb. Aggregation is important for both of them, and that is a similar degradation pathway for both of them in which you look for the increase in dimer and higher-weight molecular species. It also depends on the small molecule that is attached to the antibody. A lot of maleimides that are frequently used for conjugation can have ring openings, they can undergo reverse Michael addition under certain conditions that can cause the whole drug to fall off the antibody.“Then there is degradation of the drugs themselves. Certain classes of drugs are less stable than others. So you can have oxidation, hydrolysis, reduction reactions of the small-molecule drug, which are very difficult to pick up. If they affect the overall charge of the drug molecule, then you can see the charge heterogeneity assay. Sometimes that change is big enough such that you can pick it up with hydrophobic interaction chromatography (HIC), which often is used for the drug distribution.” |
| Stability Testing Conditions: “These typically will be very similar for ADCs and MAbs. Sometimes it will be a liquid formulation, which is typically stored at 8 °C. Other times you may have a lyophilized formulation, and it can be stored at –20 °C. But there would be no difference between an ADC and a protein in terms of storage conditions once those are identified.” |
| Potency: “One factor where there is usually a difference between a MAb and an ADC is potency. Typically, with a MAb you’re looking at binding typically through an ELISA. Many times an ADC also has to demonstrate binding, much like an ELISA. But it also has to demonstrate the ability to actually kill off the cancer cells that they bind to. Most analytical techniques remain the same for both MAbs and ADCs, but the methods themselves will typically require more development for the ADCs because of the drug component. Very hydrophobic drugs can influence size-exclusion methods and HIC analyses that are typically used to measure drug-to- antibody ratio. Charged drugs can affect the charge heterogeneity in both anion-exchange chromatography and capillary isoelectric focusing. So additional development time would need to be spent on these key stability- indicating methods.” |
| Linker: “Typically we group the linker with the drug. With a lot of ADCs, the linker–drug is actually one intermediate that is then conjugated to the antibody to create the ADC. That’s typically how it’s done. However, the Kadcyla process actually connects the linker to the antibody first and then reacts the drug to the linker.” It just depends on the ADC. The linker is also where we see some interesting degradation pathways such as the reverse Michael reaction, in which the thioether linkage between the cysteine of a MAb and the maleimide in the linker is cleaved, releasing the linker.” |
References
1 Markens U. Conducting Stability Studies: Recent Changes to Climate Zone IV. Life Science 13, March 2009.
2 Madichie C. Designing a Smart Stability Protocol for the Global Market to Minimise Time and Cost. Informa Life Sciences’ Annual Stability Testing for Pharmaceuticals and Biologics: London, UK, 25 March 2015.
3 ICH Q5C Stability Testing of Biotechnological Products. Federal Register, 10 July 1996: 36466.
4 Committee for Proprietary Medicinal Products. Note for Guidance on In-Use Stability Testing of Human Medicinal Products. The European Medinces Agency: London, UK, September 2001.
5 ICH Q1A(R2) Stability Testing of New Drug Substances and Products. Federal Register, 21 November 2003: 65717–65718.
6 ICH Q1B Stability Testing: Photostability Testing of New Drug Substances and Products. Federal Register, 16 May 1997: 27115–27122.
7 ICH Q1C Stability Testing for New Dosage Forms. Federal Register, 9 May 1997: 25634–25635.
8 ICH Q1D Bracketing and Matrixing Designs for Stability Testing of New Drug Substances and Products. Federal Register, 16 January 2003: 2339–2340.
9 ICH Q1E Evaluation of Stability Data. Federal Register, 8 June 2004: 32010–32011.
10 21 CFR 211.166. Stability Testing. Code of Federal Regulations. Food and Drug Administration: Rockville, MD, 2014.
11 Chapter <1049> Quality of Biotechnological Products: Stability Testing of Biotechnological/Biological Products. USP38–NF33. US Pharmacopeial Convention: Rockville, MD, 2014.
12 Chapter <1046> Cell and Gene Therapy Products. USP24–NF19. US Pharmacopeial Convention: Rockville, MD, 2014.
13 CPMP/QWP/609/96: Declaration of Storage Conditions. European Medicines Agency: London, UK, 2007.
14 CPMP/QWP/2934/99: In-Use Stability Testing. . European Medicines Agency: London, UK, 2001.
15 CPMP/QWP/159/96: Maximum Shelf Life for Sterile Products After First Opening or Following Reconstitution. Committe for Human Medicinal Products. European Medicines Agency: London, UK, 1998.
16 Nita AS. Outlining Key Differences in Regulatory Requirements and Expectations for Stability Studies in Key Emerging Markets. Informa Life Sciences’ Annual Stability Testing for Pharmaceuticals and Biologics: London, UK, 25 March 2015.
17 Personal correspondence with Alison Armstrong, PhD, senior director of development services at BioReliance, 2015.
18 G. Jimenez, Brake B. ICH Q5C Stability Testing of Biotechnological/Biological Products. EMA Training for ASEAN, 30–31 May 2011; www.ich.org/fileadmin/Public_Web_Site/Training/ASEAN_Q5C_workshop_May_2011/SESSION_Ia_ICH_Q5C.pdf.
19 Wonnacott K. Cell Therapy Products. FDA Office of Cellular, Tissue, and Gene Therapy, Web Seminar Series; www.fda.gov/BiologicsBloodVaccines/NewsEvents/ucm241308.htm.
20 Draft Guidance for Industry: Potency Tests for Cellular and Gene Therapy Products. US Food and Drug Administration: Rockville, MD.
21 Kling J. Highly Concentrated Protein Formulations: Finding Solutions for the Next Generation of Parenteral Biologics. BioProcess Int. 12(5) 2014: insert.
22 Waddle K, Pan W. Stability Studies in Pharmaceutical Development; www.catalent.com.
23 Panchal JP, Analyzing Subvisible Particles in Protein Drug Products: A Comparison of Dynamic Light Scattering (DLS) and Resonant Mass Measurement (RMM). Biopharmaceutical Development and Production Week, Huntington Beach, California, 31 March 2015.
24 Neergaard MS, et al. Stability of Monoclonal Antibodies at High-Concentration: Head-to-Head Comparison of the IgG1 and IgG4 Subclass. J. Pharm. Sci. 103(1) 2014: 115–127.
Maribel Rios is managing editor of BioProcess International, mrios@bioprocessintl.com.
|
Stability Testing Conference Informa’ Life Sciences Stability Testing for Pharmaceuticals and BIologics, will take place March 2016. Visit www.informa-ls.com/stabilitytesting for details. |