The goal of formulation development for therapeutic proteins is to find conditions under which a protein remains stable during storage, transport, and delivery to patients. Both chemical and physical stability must be considered. Chemical stability is related to the rates of chemical modification to a protein molecule such as deamidation of aspargine residues and oxidation of methionine residues (1, 2). Particularly important to control if they affect biological function, those modifications could also lead to changes in conformation or half-life of an antibody (3–6).
Physical stability refers to the conformational and colloidal stability of a protein. Conformational stability is its ability to maintain native secondary, tertiary, and quaternary structures. Proteins with nonnative conformation are more prone to aggregation, which can increase immunogenicity (7–9). Colloidal stability describes the tendency of native molecules to self-associate in solution, which when strong enough leads to high viscosity, phase separation, and precipitation (10–18).
All three aspects of stability are affected by how a protein is formulated. The environment it is exposed to — including factors such as pH, protein concentration, ionic strength, buffers and salts, and additional excipients — can have profound effects on all aspects of stability (10, 19). All the different aspects of stability must be considered in formulation development because a set of conditions that is favorable to one aspect of stability could compromise another. A good formulation will support stability with respect to all aspects of protein structure. That in turn supports product stability at the intended storage temperature and during product shipping and handling.
In early development of a therapeutic protein, many formulations are considered in an effort to find those that support stability. Screening a broad range of buffer conditions in parallel can minimize time and effort spent on formulation development (20, 21). The amount of available product may be limited, so an ideal screening method would require as little sample as possible.
We have devised an approach for screening formulations with samples formulated and handled primarily in 96-well microplates. The chemical, conformational, and colloidal stabilities of a protein in different formulations are assayed with methods that can be performed using minimal amounts of it with minimal handling and preparation. This approach produces identical screening plates that can be subjected to different stress conditions (e.g., incubation at elevated temperature, agitation, and exposure to light).
We used data from a formulation screening to select lead formulations that we then prepared on a larger scale to monitor in accelerated and real- time stability studies. Comparing results from the screening study with a subsequent stability study suggested that some assays (e.g., solubility) were predictive of long-term stability whereas others (e.g., differential scanning fluorimetry, DSC) were not.
Materials and Methods
Antibody Samples: Fully human IgG1 antibody was produced by Chinese hamster ovary (CHO) cells grown in 1-L to 10-L bioreactors using a proprietary chemically defined medium and feeds. Cells were removed from the culture suspension by depth filtration, and antibodies were purified from the supernatant using sequential protein A affinity, anion-exchange, and cation-exchange chromatography. After purification, the antibodies were formulated in 20 mM sodium citrate at pH 6.0 with 150 mM NaCl and stored at 2–8 °C until needed. Concentration of the purified antibody samples was 10–15 mg/mL.
Buffers and Reagents: We purchased USP- or ACS-grade (US Pharmacopeia and American Chemical Society) chemicals from Sigma-Aldrich Fine Chemicals and performed dialysis using Slide-a-Lyzer cassettes (Life Technologies) with 10-kDa cutoff. We bought Sypro Orange protein dye from Life Technologies. Formulation buffers were prepared as 200mM stock solutions using previously defined ratios of acidic and basic salts for each buffer. We added sodium chloride (NaCl) to the formulated antibody samples from a 3M stock.
Formulation for Screening: Purified antibody was dialyzed twice at room temperature against water using a volume of water >100× the sample volume. We measured concentration after dialysis and adjusted it to 5.5 mg/mL using water as a diluent. Then we dispensed 100 µL of the 200 mM buffer solutions into wells of a non–protein-binding ScreenMate polypropylene deep-well plate (Matrix Technologies). Next, we added 900 µL of the dialyzed antibody solution into wells using a multichannel pipette for a 1-mL solution containing 5 mg/mL antibody and 20 mM buffer. We added NaCl to some wells with 30 µL of a 3M solution to a final concentration of 148 mM.
From those deep-well–plate solutions, we transferred 200 µL to corresponding wells of four standard 96-well polystyrene Nunc Microwell plates (Thermo Scientific), as in Figure 1. We sealed those plates with MicroMat silicone plate-sealing mats (SunSRI), then incubated one plate at 4 °C for four weeks, one at 37 °C for two weeks, and one at 37 °C for four weeks. We assayed samples from the fourth plate on the same day the plates were prepared.
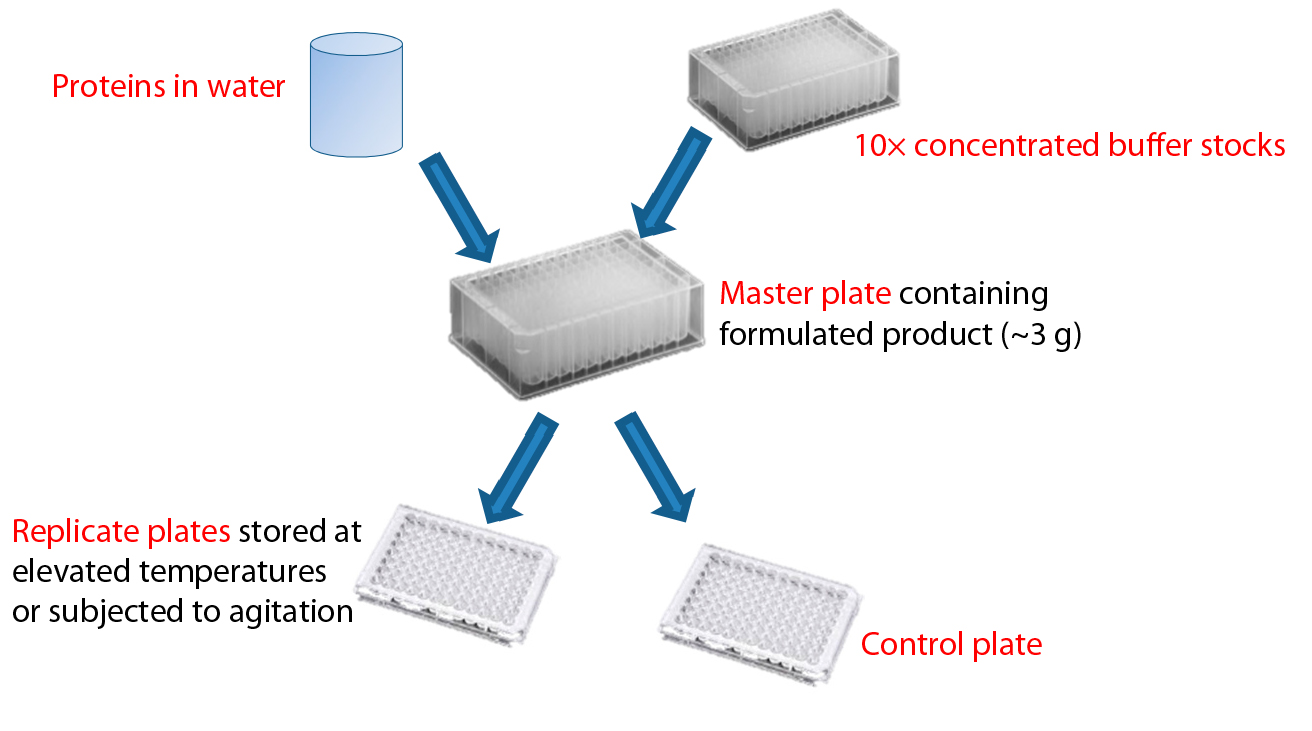
Figure 1: Formulation of proteins using concentrated buffer stocks; protein that has been dialyzed into water is mixed with a concentrated buffer to create a master plate with the desired formulation conditions. Replicate plates created from the master plate allow comparison of stability to different stress conditions using identical formulations.
Assessment of Evaporation from 96-Well Plates: To determine the volume of liquid lost through evaporation during incubation at 37 °C, we filled the wells of a microplate with 200 µL of a phenol red solution with an absorbance of 0.7 at 440 nm, then immediately read their absorbance using a Spectramax M5 plate reader. Then that plate was sealed with a sealing mat and incubated at 37 °C. We measured its absorbance every three to four days for four weeks and calculated the change in volume from the resulting path-length–corrected values.
Differential Scanning Fluorimetry: For DSF, we diluted formulated antibody samples in matching buffers to 0.2 mg/mL, then mixed 20 µL samples with 20 µL of a 1:500 dilution of Sypro Orange dye in the wells of a MicroAmp thin-walled 96-well plate from ABI (22). For this assay, we used an ABI 7300 real-time polymerase chain reaction (RT-PCR) instrument. It was programmed to increase temperature in 1 °C increments, each followed by a 1-min. hold before measurement of Sypro Orange fluorescence at 576 nm. We calculated the temperature of transition from a native to an unfolded state (TH) using a modified version of ThermoQ software that uses a four- parameter logistic curve-fit equation for fluorescence and temperature data (23). The C parameter is the temperature at the inflection point of a plot of the rising fluorescence:
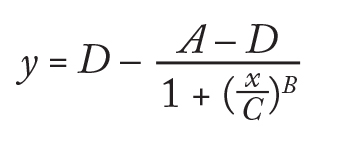
Size-Exclusion UPLC: We determined the levels of soluble high–molecular- weight (HMW) species with a 4.6 × 150 mm Waters Acuity BEH 200 column on a Waters H-class UPLC with a flow-through needle injector and tunable ultraviolet (TUV) absorbance detector. The mobile-phase buffer contained 20 mM sodium phosphate at pH 7.0 with 300 mM NaCl and 0.02% sodium azide and flowed at 0.5 mL/min. We diluted formulated samples to 1 mg/mL in mobile-phase buffer for analysis.
Absorbance (340 nm): We assessed antibody aggregationy measuring absorbance at 340 nm of wells containing 200 µL of formulated antibody with a Versamax plate reader (Molecular Devices). A clear UV Star low-UV absorption plate (Greiner Bio One) minimized background absorbance.
Sypro Orange Binding: After measuring absorbance at 340 nm, we transferred a 50-µL aliquot of solution from each well to a clean 96-well solid black assay plate. Then we added 50 µL of Sypro Orange dye diluted 1:500 in water to all wells. Control wells contained formulation buffers without antibody. We measured fluorescence (ex 485 nm, em 590 nm) with a Victor 3 plate reader (Perkin Elmer).
Solubility: High protein solubility correlates with high colloidal stability (17, 24). Solubility can be assessed in different ways, but one simple method is to use a precipitant — e.g., polyethylene glycol (PEG) or ammonium sulfate — to create conditions that simulate very high protein concentrations by crowding protein molecules or withdrawing water from them (25). Our approach was to add to all formulations a single concentration of precipitant (ammonium sulfate) that is sufficient to cause partial precipitation of antibody and then to measure the concentration of the remaining soluble antibody (26, 27). Then we could rank antibody solubility in each buffer relative to the others.
First, we empirically determined the concentration of ammonium sulfate required to precipitate half the test antibody formulated in sodium citrate buffer at pH 6 to be 1.2 M. Next, we prepared a 2.4 M ammonium sulfate solution and mixed it with an equal volume of formulated antibody. After overnight incubation at room temperature, precipitated protein was removed by a 0.2-µm polyvinylidene fluoride (PVDF) filter plate so we could measure the concentration of soluble protein by absorbance at 280 nm using a Nanodrop spectrophotometer. To better maintain pH in the presence of high ammonium-sulfate concentrations, we used 50 mM buffers in the solubility experiments (Figure 7) rather than 20 mM.
Capillary Gel Electrophoresis: Our Beckman-Coulter ProteomeLab PA 800 capillary electrophoresis system is equipped with a UV absorbance detector. Denaturing sample buffer and a sieving gel matrix came with a ProteomeLab SDS-MW analysis kit from Sciex. We diluted formulated samples to 1 mg/mL with sample buffer containing either 5% β-mercaptoethanol (for reduced samples) or 12.5 mM sodium iodoacetate (for nonreduced samples). To denature the samples, we heated them to 70 °C for 10 minutes, cooled them to room temperature, and then injected them into a fused-silica capillary filled with the sieving gel matrix. Separation at 25 °C took 30 minutes at 15 kV, and antibodies were detected through their absorbance at 214 nm.
Capillary Isoelectric Focusing (cIEF): After desalting formulated samples by ultrafiltration using Amicon Ultra centrifugal filters (EMD Millipore), we adjusted protein concentration to 0.3 mg/mL in an ampholyte solution containing 4.8% Pharmalyte 3–10 (GE Healthcare), 40 mM arginine, 1.6 mM iminodiacetic acid, and four synthetic peptide pI markers (Sciex). We filled a neutral-coated Sciex separation capillary with the sample/ ampholyte solution, then carried out cIEF at 30 kV for 15 minutes. The anode buffer was phosphoric acid, and the cathode buffer was sodium hydroxide. Focused proteins were mobilized by changing anode and cathode buffers to 300 mM acetic acid, and antibodies were detected through their absorbance at 280 nm.
Confirmation of the Screening Results with Traditional Stability Study: We used six candidate formulation buffers (selected through high- throughput screening) to compare the 96-well method for formulation and screening with a traditional method of dialysis against several buffers and a longer-term accelerated stability study. After concentration to 30 mg/mL by diafiltration, the purified antibody was formulated by dialysis against each buffer, then dispensed into 3-mL borosilicate vials with silicone stoppers. We stored inverted vials at 37 °C and at 4 °C for up to six months. At each time point, we removed vials from storage and analyzed samples with SE-UPLC, isoelectric focusing, and capillary gel electrophoresis of both reduced and nonreduced samples.
Results
Assessment of Evaporation from 96-Well Plates: Incubation at 37 °C can lead to significant evaporation from wells if microplates are not adequately sealed. We found a silicone plate sealing mat to be an effective means of minimizing evaporation for up to a month. Table 1 lists volume changes in a sealed plate after one-month incubation at 37 °C, indicating that over 30 days the volume decreased 6–10% in the central 60 wells of the plate, 10–15% from wells on the plate edges, and 20% from corner wells. Evaporation of 10% corresponds to an 11% increase in buffer, NaCl, and protein concentration in a formulation screen. To minimize those evaporation effects, we filled the edge wells with water and used only the center 60 wells of the plate for formulated protein.
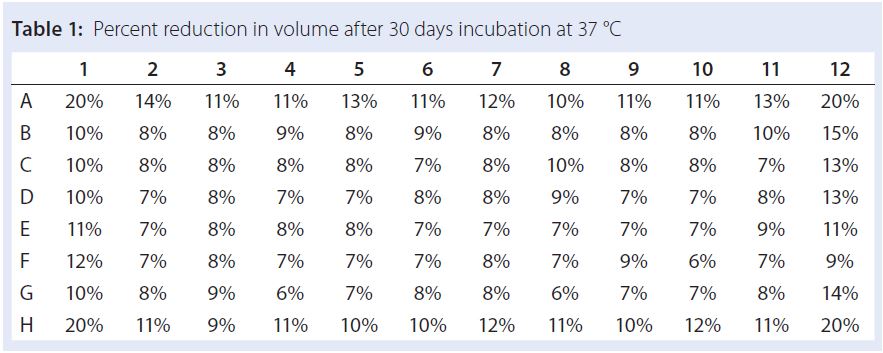
Table 1: Percent reduction in volume after 30 days incubation at 37 °C
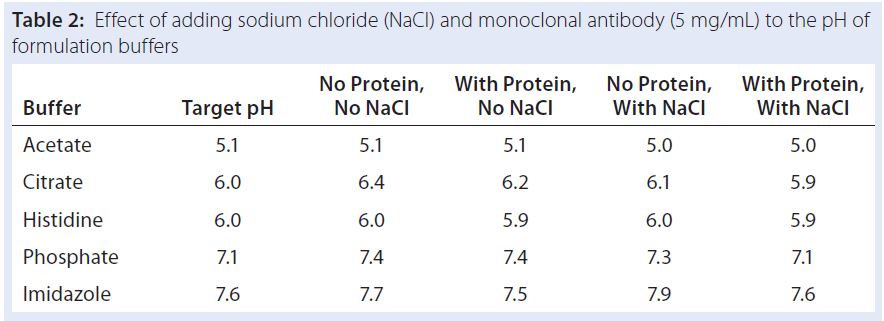
Table 2: Effect of adding sodium chloride (NaCl) and monoclonal antibody (5 mg/mL) to the pH of formulation buffers
Being zwitterionic, proteins can act as pH buffers and might change the pH of their formulation buffers. Highly concentrated antibody solutions have significant buffering capacity in the pH 4–6 range (28). So we tested the pH of some formulated antibody solutions and compared it with the pH of buffers without antibody. Table 2 shows that addition of antibody at 5 mg/mL lowered the pH of formulation buffers by 0.1–0.3 units, but the pH of the sodium acetate formulation buffer at pH 5 did not change. Those small changes in pH after antibody addition are consistent, suggesting that the antibody does lower pH slightly.
Experimental Design: Our approach was to focus on formulation buffers that (based on prior knowledge) are most likely to provide maximum stability for human MAbs. However, this same experimental approach can be applied to proteins that are stable under a different range of conditions or other cases in which investigators wish to screen a larger range. We selected our pH buffers based on their buffering capacity in the 4–8 pH range, in which human MAbs are most stable. Except for sodium succinate and imidazole-HCl, these buffers are used to formulate at least one licensed therapeutic monoclonal antibody (according to drugs@FDA data files online at www.fda.gov/ Drugs/InformationOnDrugs/ ucm079750.htm).
We prepared each buffer as a 200 mM stock solution without NaCl, using predefined ratios of a weak acid and corresponding base (or free base and HCl for imidazole and histidine buffers). Each formulation buffer was evaluated at three pH values both with and without 148 mM NaCl. Predefined ratios of acid and base had been determined empirically to produce the desired pH values. Use of such ratios eliminates the need to adjust the pH of stock solutions, a step that could otherwise contribute to variability.
Figure 1 depicts our approach to preparing replicate microplates with identical formulations. The antibody to be formulated is first dialyzed against water to minimize carryover of purification buffers, then added to 10× concentrated buffers arranged in a 96– deep-well plate for a final buffer concentration of 20 mM and NaCl concentration(when present) of 148 mM. Excipients such as nonionic detergents, sugars, or sugar alcohols are added as concentrated stock solutions at this point. Up to five identical replicate plates then can be made from that deep-well plate. After immediate analysis of samples in one replicate plate, the other plates are subjected to stress conditions such as elevated temperature or agitation before analysis. This analytical work flow was designed to simplify sample handling and make efficient use of available material (see the “Work Flow” box).
Platform Work Flow
| Absorbance of solutions at 340 nm is measured in all wells using a 96-well plate reader to assess the levels of insoluble aggregates. |
| A portion of each solution is diluted to 1 mg/mL with SE-UPLC mobile phase buffer in a 96-well plate and subjected to SE-UPLC analysis to assess soluble HMW species (SE-UPLC). |
| Another portion of the formulations is diluted in their corresponding buffers, mixed with the environment-sensitive Sypro Orange fluorescent dye, and then analyzed by DSF. SYPRO Orange dye has low fluorescence in aqueous solution, but when bound to the hydrophobic regions of thermally denatured proteins it increases in fluorescence significantly (29, 30). The DSF assay uses a 96-position RT-PCR thermocycler. |
| Another Sypro Orange assay is used to measure surface hydrophobicity of the antibody at room temperature (31, 32). After mixing it with 20 µL of undiluted antibody, we measure fluorescence with a plate reader to observe protein conformational changes and aggregation (33). |
| Finally, a portion of the remaining solutions is mixed with an ammonium sulfate solution and incubated overnight. The concentration of ammonium sulfate causes precipitation of ~50% of the antibody. (Ammonium sulfate binds water molecules more tightly than proteins do, thus reducing available water available to solubilize protein.) After incubation, we measure the concentration of soluble protein using a Nanodrop spectrophotometer. This method provides information about the relative solubility of proteins in different formulation buffers (27). |
Case Study
We studied the effects of different buffers, NaCl, and formulation pH on a purified human IgG1 MAb.
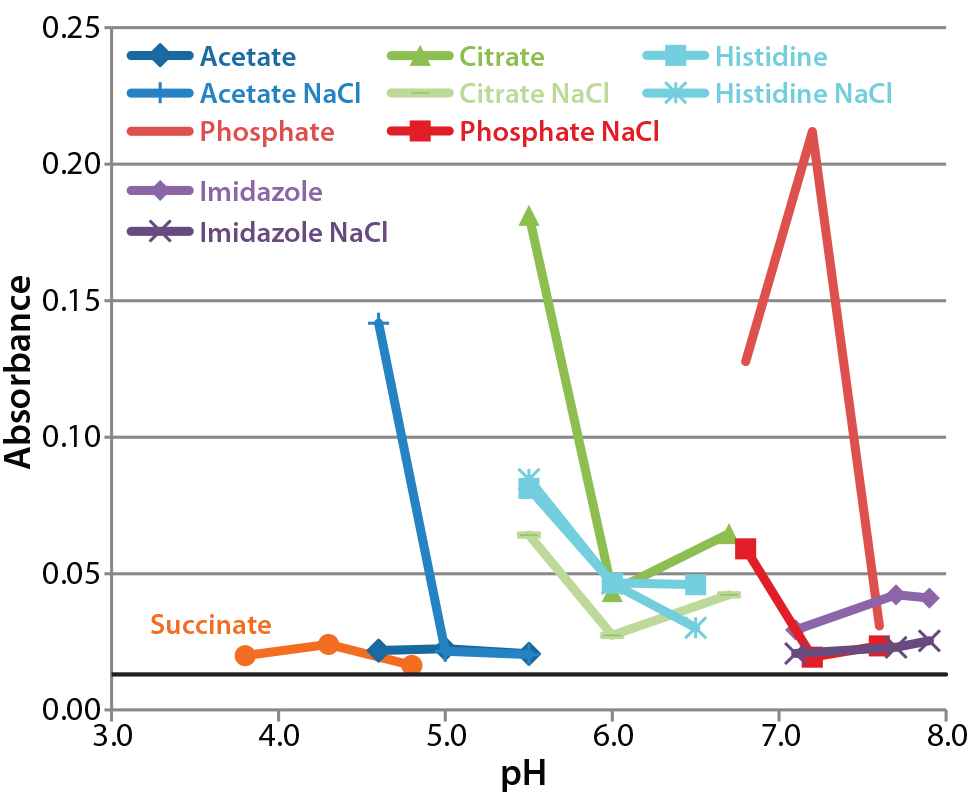
Figure 2: A340 after 30-day incubation at 37 °C; data for sodium succinate buffer in the presence of NaCl are omitted because their high values would impair the clarity of the other data points. Absorbance at the initial time point was 0.013 (solid black line), with a range of 0.009–0.017.
Assessment of Protein Aggregation By 340-nm Absorbance: Figure 2 presents absorbance data at 340 nm after 28 days incubation of each formulation at 37 °C. For most formulations, absorbance increased slightly during incubation, increasing to >1.0 in two of the three formulations containing sodium succinate and NaCl. Absorbance of succinate formulations without NaCl was much lower.
Other formulations with high absorbance contained acetate buffer with NaCl at pH 4.6, citrate buffer at pH 5.5, and phosphate buffer at pH 6.6 and 7.1. The high absorbance values obtained with these formulations reflect the level of aggregation occurring, but it is hard to discern a pattern in the conditions that cause it. We found the smallest increase in absorbance over time in acetate and succinate formulations without NaCl and the largest increases in phosphate, citrate, and histidine buffers. NaCl generally increased aggregation with succinate buffers, but at pH >6.8, it appeared to decrease the amount of aggregation.
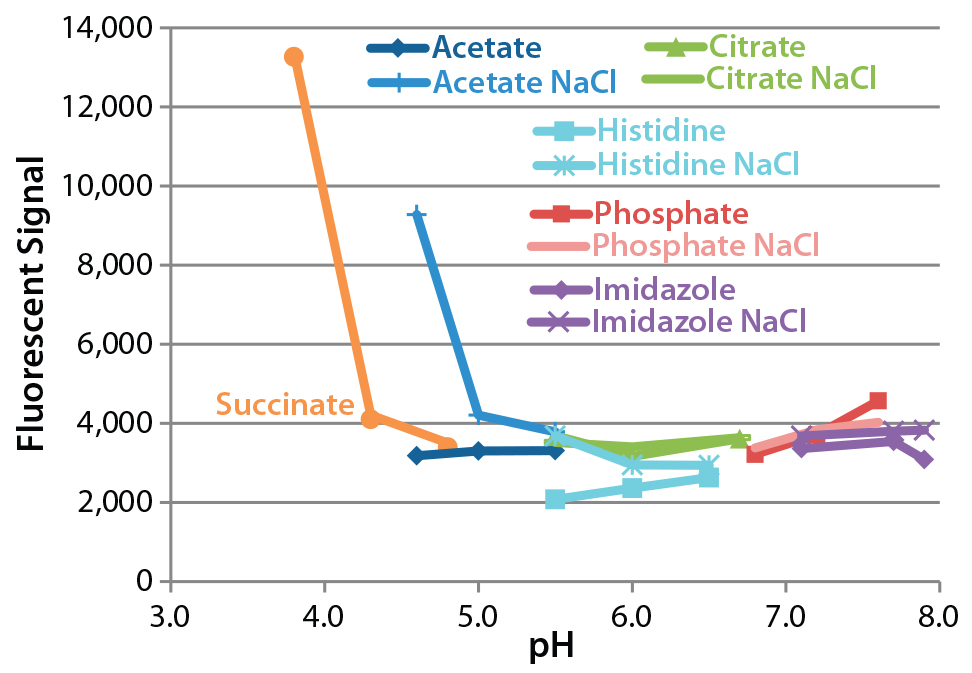
Figure 3: Sypro Orange fluorescence after 28-day incubation of monoclonal antibody (MAb) at 37 °C; data for sodium succinate buffer in the presence of NaCl are omitted because their high values would impair the clarity of the other data points. The average signal immediately after formulation was 3,338, with a coefficient of variation of 8% and a range of 2,944–3,724.
Assessment of Protein Hydrophobicity and Aggregation By Sypro Orange Binding Assay: Figure 3 shows the Sypro Orange binding assay results, in which an increase in fluorescence indicates either aggregation or a conformational change in the antibody. Fluorescence greatly increased after 28 days at 37 °C in formulations with a pH <5.0, particularly those using succinate as a buffer. The presence of NaCl had little effect except increasing fluorescence at low pH. These results suggest that incubation at 37 °C increases the exposure of hydrophobic regions of this particular antibody. Interestingly, the A340 and Sypro Orange results both suggest that its formulation either at low pH (<5.0) or in a succinate buffer would decrease stability.
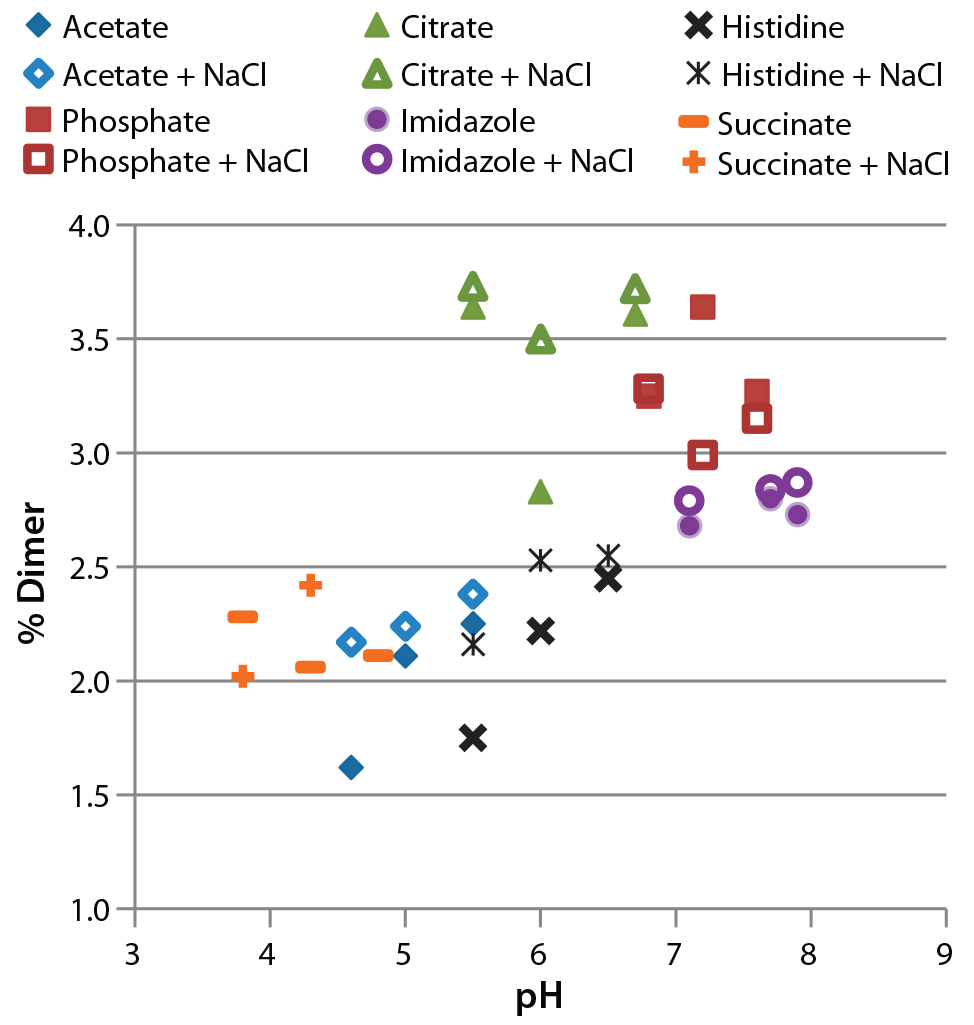
Figure 4: Analysis of formulated antibody by size-exclusion ultraperformance liquid chromatography (UPLC); samples were incubated for four weeks at 37 °C before analysis. The percentage of dimer (1.2% immediately after formulation) is plotted against pH.
SE-UPLC to Assess Protein Aggregation: Size-exclusion UPLC can resolve soluble antibody aggregates as large as tetramers as well as antibody fragments. Figure 4 plots the relative amount of dimeric antibody in each condition. Under 0.4% fragments and HMW forms were observed. There is a clear trend of increasing pH causing higher dimer levels. For example, formulation in citrate buffer (green) resulted in a higher dimer level than formulation in acetate or histidine buffers of similar pH. The effects of NaCl on dimerization were small, with no clear trends in the data except that adding it to the histidine buffer increased dimer levels.
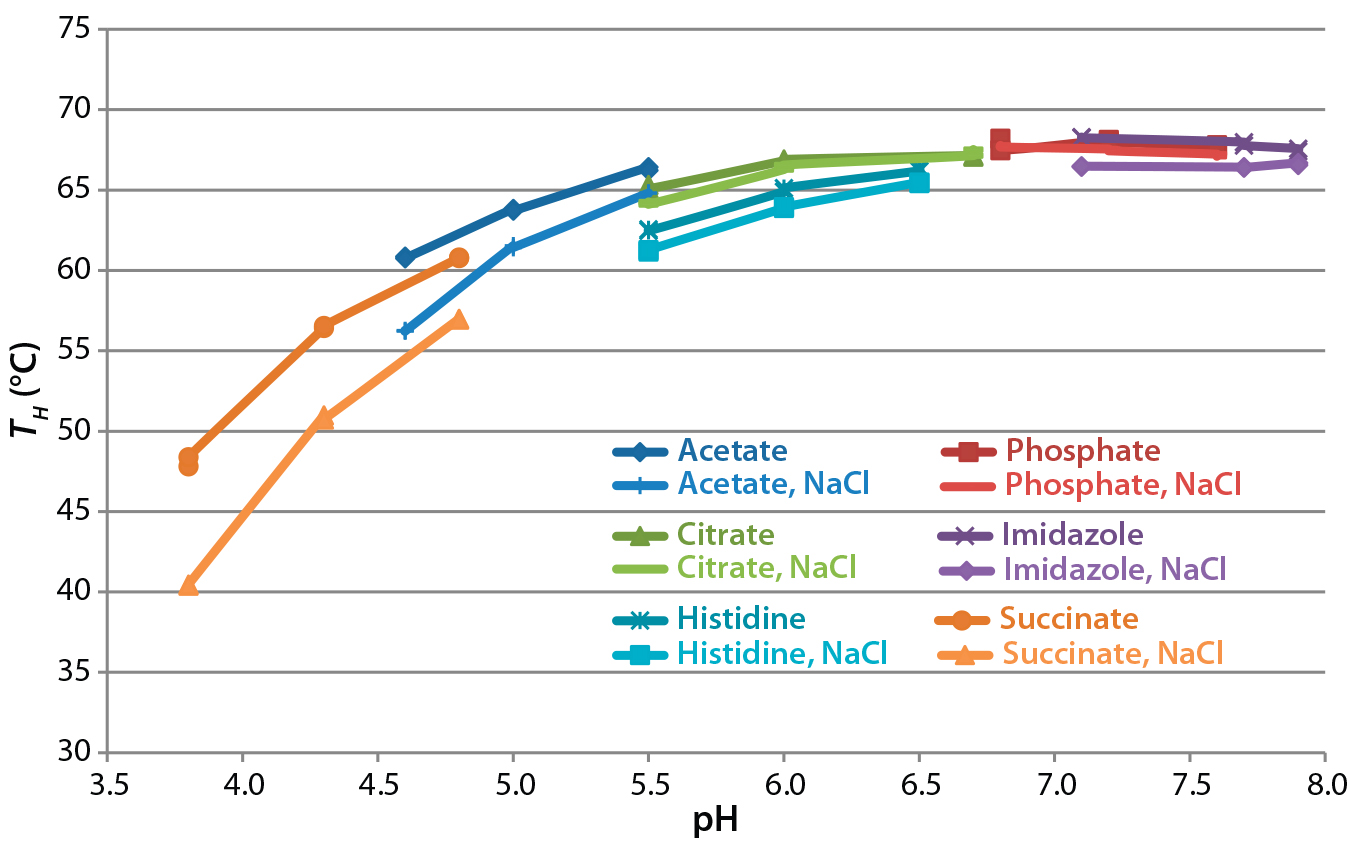
Figure 5: Analysis of formulated antibody by differential scanning fluorimetry with Sypro Orange protein gel dye; the temperature of hydrophobic exposure (TH) was determined by monitoring fluorescence as temperature was increased from 40 °C to 95 °C and fitting a four-parameter logistic curve to the resulting data.
Assessment of Protein Thermal Stability: We performed DSF using Sypro Orange dye as a means to examine the thermal stability of the antibody in different conditions. Figure 5 compares the temperature of hydrophobic exposure (TH) in all buffers. TH increases with pH between 3.8 and 6.5 and is independent of it above pH 6.5. Different buffer substances had some effect at pH <6.5, with acetate buffers conferring greater thermal stability than histidine, citrate, or succinate buffers. NaCl reduced TH at pH values <6.5 but had no significant effect >pH 6.5. DSF results suggested that the greatest stability would be at pH >6.5.
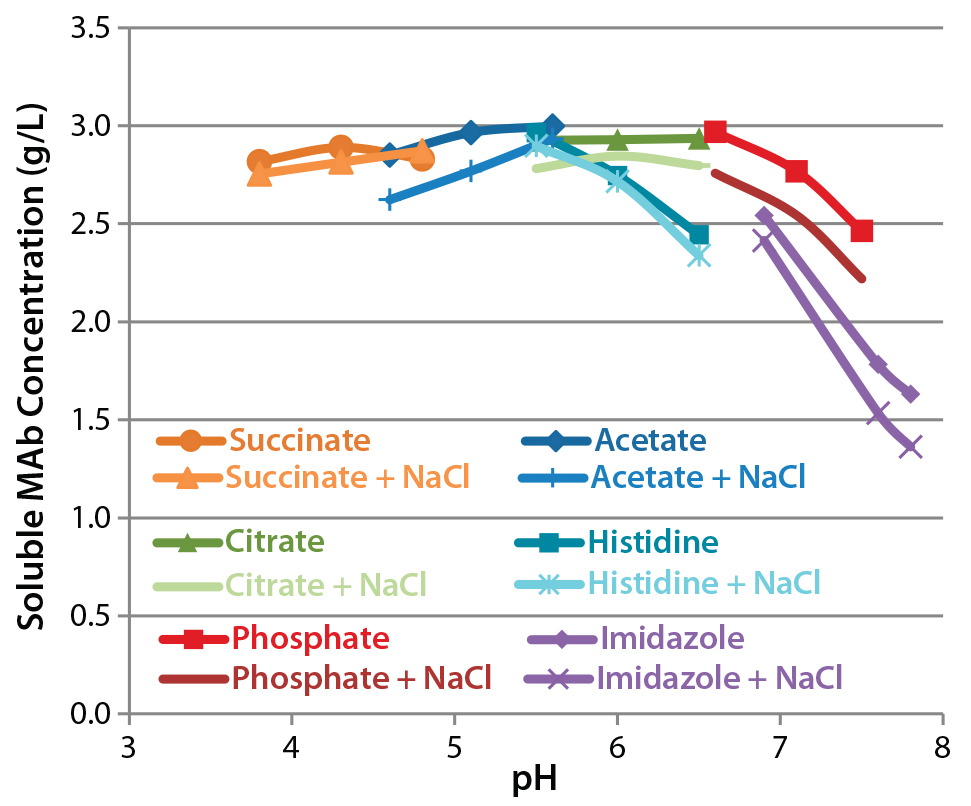
Figure 6: Solubility in 1.2 M ammonium sulfate; equal volumes of a 5-mg/mL antibody formulation and 2.4 M ammonium sulfate were mixed and incubated overnight. Precipitated protein was removed by centrifugation, and the concentration of soluble protein was determined by absorbance.
Assessment of Protein Solubility: Figure 6 depicts the relationships between pH, NaCl, and solubility. At pH ≤6, antibody solubility is similar in different buffers. As pH rises, solubility decreases as some differences between formulations become clear. Citrate- and phosphate-buffered formulations conferred greater solubility than histidine- and imidazole-buffered formulations. NaCl in general reduced solubility under these conditions but to a lesser extent than did high pH.
These assay results illustrate the balance that must be made in choosing a formulation. No single formulation provides optimal conditions for protein solubility as well as chemical, conformational, and colloidal stability. The SE-UPLC and solubility results suggest that a lower formulation pH will provide greater stability to the antibody (with lower dimer percentage and higher solubility). On the other hand, DSF results suggest that a higher formulation pH provides greater stability with higher TH.
To investigate those differences, we divided the antibody into six different buffers by dialysis: sodium acetate pH 5.7, sodium citrate pH 6, and sodium phosphate pH 7.2, each in the absence or presence of 150 mM NaCl. Antibody concentration was adjusted to 25 mg/mL, polysorbate 80 was added to 0.025%, and the resulting solution was filled into borosilicate glass vials with silicone stoppers. Inverted vials were incubated at 4 °C and 37 °C for up to six months, with 1-mL aliquots periodically taken aseptically and analyzed by SE-UPLC, capillary sodium-dodecyl sulfate (SDS) gel electrophoresis (CGE), and isoelectric focusing.
CGE results from samples incubated for six months at 37 °C indicated that antibody integrity was greatest in an acetate-buffer formulation without NaCl and lowest in sodium phosphate buffers (data not shown). The cIEF assay demonstrated that antibody formulated in sodium acetate or sodium citrate buffers was more stable than that formulated with sodium phosphate. And the SE-UPLC results (in agreement with those assays) showed that the level of dimeric antibody is lowest in sodium acetate buffers.
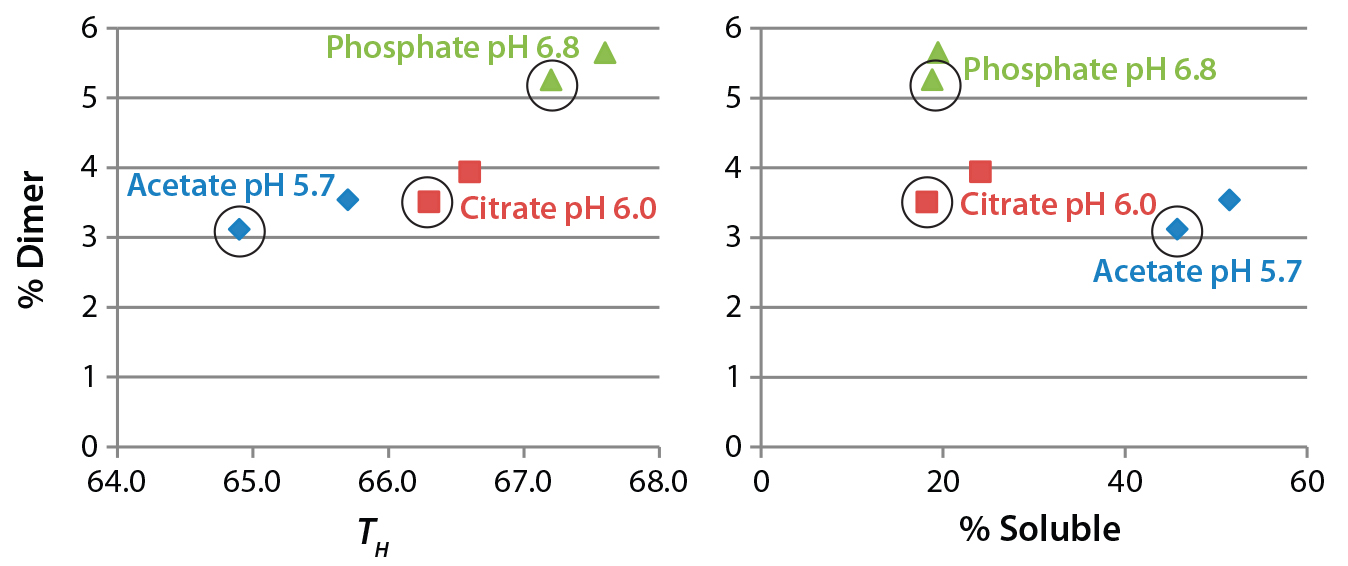
Figure 7: Comparing the level of antibody dimer in formulations incubated at 37 °C for six months with differential scanning fluorimetry (left) and solubility results (right)
Figure 7 compares the percent dimer measured in six-month accelerated stability samples with TH in a DSF assay or fraction of soluble antibody in the presence of 1.2 M ammonium acetate. In the DSF assay, higher melting temperature actually correlated with higher dimer levels, whereas higher solubility (a greater fraction remaining soluble) correlated with lower dimer levels in the accelerated stability samples. The DSF assay therefore does not seem to be a useful method for predicting formulations that minimize soluble aggregates. That is surprising because the method is used commonly in formulation screening (22). Analysis of additional antibody samples will be needed to determine the generality of our conclusion.
Discussion
We have presented a flexible, high- throughput approach for screening a large number of formulation buffers in a limited time with a small quantity of antibody to find those in which protein stability is maximized. Key features of this method are its use of concentrated formulation buffers prepared with predefined ratios of acid and base components, preparation of replicate 96-well plates that can be treated with different stress conditions, and use of sensitive assays that require minimal samples.
Buffer Stocks: Our use of concentrated buffer stocks eliminated the need for buffer exchange into many different buffers, which saved both time and material. Only the antibody itself is buffer-exchanged into water before addition to concentrated buffers. Other dilute buffers could be used instead of water. Our screening data show that pH is important to stability, so pH measurements would have to be made after adding a buffered protein to concentrated formulation buffers.
To increase the consistency of results, we prepared buffers according to formulas defined during initial development of this method (without any pH adjustment after buffer preparation). They were designed using the Henderson–Hasselbach equation and a temperature-compensated buffer pKa to target a particular pH value. We measured pH after diluting concentrated buffers based on those formulas, assigning the measured pH to each buffer. Adding NaCl and protein affects solution pH by changing ionic strength or introducing a buffer substance (or the protein itself), so we measured those changes and found them to be small (Table 2).
Replicate Microplates: Creating a master plate with ~1 mL of each formulation allows several replicate plates to be produced and subjected to different treatments. Our experiments focused on elevated- temperature incubation, but the same technique could be used to study stability with agitation, light, or freezing.
Microplates simplify handling in all these assays. The least sensitive test (absorbance at 340 nm) requires the largest sample volume but is nondestructive: 200 µL of a 5 mg/mL antibody solution is sufficient, but smaller volumes or lower concentrations could be used if treatment creates high levels of insoluble aggregate (e.g., vigorous agitation). The Sypro Orange binding assay uses 20 µL of undiluted formulation, and DSF and SE-UPLC each require 20 µL of <1-mg/mL sample. Dilutions for each assay are made in the same plate used for that assay. The solubility assay also could be performed with 96-well filter plates, a UV transparent collection plate, and a plate reader with UV capability. For the formulation conditions described herein, a total of 200 mg of protein was needed for five replicate plates, each tested using these assays.
The solubility assay format is novel. Rather than performing serial dilutions of the precipitant for each formulation buffer, we tested all buffers at 1.2 M ammonium sulfate. This gave different concentrations of soluble antibody, dependent on the formulation. Although the resulting data could not be used to predict maximum antibody solubility in a particular buffer, it provided sufficient information to rank buffers with regard to their ability to maintain solubility. For a given antibody, solubility should be lowest at its isoelectric point (pI 8.5 in this case), so it would be expected to decrease as formulation pH approaches the antibody’s pI.
Comparing Results: The results of different assays can lead to different conclusions regarding an optimal formulation. Our results indicate the need to balance protein stability as measured by different methods. DSF assay results suggested that pH >6.5 is optimal, whereas SE-UPLC and solubility assays suggested that pH <6.5 is preferred. That disagreement must be attributable to the properties measured by each assay.
DSF measures antibody unfolding as temperature increases, and stabilization against unfolding at high temperatures is thought to correlate with greater conformational stability at lower temperatures. Greater conformational stability in turn would limit the level of nonnative molecules and their aggregation. SE-UPLC measures antibody self-interaction, but native or nonnative molecules can participate in such interactions. For example, native molecules could interact through hydrophobic or electrostatic means. The solubility assay measures colloidal stability of native antibody, assuming that the vast majority of molecules present are in their native state.
According to our data, stabilization at high temperatures by formulation pH >6.5 does not correlate with stability of this antibody at lower temperatures (27). The antibody we used has a TH of 65–70 °C, roughly 30 °C above the temperature in our six-month stability study. DSF assay results might be more predictive of stability if TH was closer to the intended storage temperature. We will have to study more proteins to gain a better understanding of the relationship between DSF unfolding temperature and real-time stability.
By contrast, the utility of the solubility assay was confirmed by comparing results of a six-month accelerated stability study with the screening results. The solubility assay accurately predicted the buffers with which dimer levels would be lowest. The same formulations that supported the highest solubility also provided good chemical stability as judged with CGE and IEF. Again, studies of additional MAbs will be needed to support the generality of these conclusions.
The assays used herein primarily measure physical and colloidal stability. The usefulness of this and similar screening platforms will be improved by including high- throughput assays to measure chemical degradations that commonly occur during storage of protein therapeutics.
References
1 Liu H, et al. Heterogeneity of Monoclonal Antibodies. J. Pharm. Sci. 97(7) 2008: 2426–2447.
2 Davies MJ. The Oxidative Environment and Protein Damage. Biochim. Biophys. Acta. 1703(2) 2005: 93–109.
3 Yan B, et al. Analysis of Post- Translational Modifications in Recombinant Monoclonal Antibody IgG1 By Reversed-Phase Liquid Chromatography/Mass Spectrometry. J. Chromatogr. A 1164(1–2) 2007: 153–161.
4 Luo Q, et al. Chemical Modifications in Therapeutic Protein Aggregates Generated Under Different Stress Conditions. J. Biol. Chem. 286(28) 2011: 25134–25144.
5 Haberger M, et al. Assessment of Chemical Modifications of Sites in the CDRs of Recombinant Antibodies: Susceptibility vs. Functionality of Critical Quality Attributes. MAbs 6(2) 2014: 327–339.
6 Yan B, et al. Succinimide Formation at Asn 55 in the Complementarity Determining Region of a Recombinant Monoclonal Antibody IgG1 Heavy Chain. J. Pharm. Sci. 98(10) 2009: 3509–3521.
7 Brummitt RK, Nesta DP, Roberts CJ. Predicting Accelerated Aggregation Rates for Monoclonal Antibody Formulations, and Challenges for Low-Temperature Predictions. J. Pharm. Sci. 100(10) 2011: 4234–4243.
8 Joubert MK, et al. Highly Aggregated Antibody Therapeutics Can Enhance the In Vitro Innate and Late-Stage T-Cell Immune Responses. J. Biol. Chem. 287(30) 2011: 25266– 25279. 9 Rombach-Riegraf V, et al. Aggregation of Human Recombinant Monoclonal Antibodies Influences the Capacity of Dendritic Cells to Stimulate Adaptive T-Cell Responses In Vitro. PLoS One 9(1) 2014: e86322.
10 Lewus RA, et al. Interactions and Phase Behavior of a Monoclonal Antibody. Biotechnol. Progr. 27(1) 2011: 280–289.
11 Lewus RA, et al. A Comparative Study of Monoclonal Antibodies. 1: Phase Behavior and Protein–Protein Interactions. Biotechnol. Progr. 31(1) 2015: 268–276.
12 Liu J, et al. Reversible Self-Association Increases the Viscosity of a Concentrated Monoclonal Antibody in Aqueous Solution. J. Pharm. Sci. 94(9) 2005: 1928–1940.
13 Mason BD, et al. Opalescence of an IgG2 Monoclonal Antibody Solution As It Relates to Liquid–Liquid Phase Separation. J. Pharm. Sci. 100(11) 2011: 4587–4596.
14 Salinas BA, et al. Understanding and Modulating Opalescence and Viscosity in a Monoclonal Antibody Formulation. J. Pharm. Sci. 99(1) 2010: 82–93.
15 Sule SV, et al. High-Throughput Analysis of Concentration-Dependent Antibody Self- Association. Biophys. J. 101(7) 2011: 1749–1757.
16 Trilisky E, et al. Crystallization and Liquid–Liquid Phase Separation of Monoclonal Antibodies and Fc-Fusion Proteins: Screening Results. Biotechnol. Progr. 27(4) 2011: 1054–1067.
17 Wang Y, et al. Quantitative Evaluation of Colloidal Stability of Antibody Solutions Using PEG-Induced Liquid–Liquid Phase Separation. Mol. Pharm. 11(5) 2014: 1391–1402.
18 Wang Y, et al. Phase Separation in Solutions of Monoclonal Antibodies and the Effect of Human Serum Albumin. Proc. Natl. Acad. Sci. USA 108(40) 2010: 16606–16611.
19 Wang W, et al. Antibody Structure, Instability, and Formulation. J. Pharm. Sci. 96(1) 2007: 1–26.
20 Capelle MA, Gurny R, Arvinte T. High Throughput Screening of Protein Formulation Stability: Practical Considerations. Eur. J. Pharm. Biopharm. 65(2) 2007: 131–148.
21 Senisterra GA, Finerty PJ Jr. High Throughput Methods of Assessing Protein Stability and Aggregation. Molec. Biosyst. 5(3) 2009: 217–523.
22 He F, et al. High Throughput Thermostability Screening of Monoclonal Antibody Formulations. J. Pharm. Sci. 99(4) 2010: 1707–1720.
23 Phillips K, de la Pena AH. Chapter 10: The Combined Use of the Thermofluor Assay and ThermoQ Analytical Software for the Determination of Protein Stability and Buffer Optimization As an Aid in Protein Crystallization. Current Protocols in Molecular Biology. Ausubel FM, et al., Eds. John Wiley and Sons: Hoboken, NJ, 1995.
24 Gibson TJ, et al. Application of a High- Throughput Screening Procedure with PEG- Induced Precipitation to Compare Relative Protein Solubility During Formulation Development with IgG1 Monoclonal Antibodies. J. Pharm. Sci. 100(3) 2011: 1009–1021.
25 Kramer RM, et al. Toward a Molecular Understanding of Protein Solubility: Increased Negative Surface Charge Correlates with Increased Solubility. Biophys. J. 102(8) 2012: 1907–1915.
26 Trevino SR, Scholtz JM, Pace CN. Measuring and Increasing Protein Solubility. J. Pharm. Sci. 97(10) 2008: 4155–4166.
27 Banks DD, et al. Native-State Solubility and Transfer Free Energy As Predictive Tools for Selecting Excipients to Include in Protein Formulation Development Studies. J. Pharm. Sci. 101(8) 2012: 2720–2732.
28 Gokarn YR, et al. Self-Buffering Antibody Formulations. J. Pharm. Sci. 97(8) 2008: 3051–3066. 29 Pantoliano MW, et al. High-Density Miniaturized Thermal Shift Assays As a General Strategy for Drug Discovery. J. Biomol. Screen 6(6) 2001: 429–440.
30 Ericsson UB, et al. Thermofluor-Based High-Throughput Stability Optimization of Proteins for Structural Studies. Analyt. Biochem. 357(2) 2006: 289–298.
31 Cardamone M, Puri NK. Spectrofluorimetric Assessment of the Surface Hydrophobicity of Proteins. Biochem. J. 282(2) 1992: 589–593.
32 Cheng W, et al. Linking the Solution Viscosity of an IgG2 Monoclonal Antibody to Its Structure As a Function of pH and Temperature. J. Pharm. Sci. 102(12) 2013: 4291– 4304.
33 He F, et al. Detection of IgG Aggregation By a High Throughput Method Based on Extrinsic Fluorescence. J. Pharm. Sci. 99(6) 2010: 2598–2608.
Paul Casaz is a research scientist, Anna Brousseau is a research associate, and Sadettin Ozturk is deputy director of process development with Massbiologics at the University of Massachusetts Medical School, 460 Walk Hill Street, Boston, MA 02126; 1-617-474-3000; www.umassmed. edu/massbiologics.