Assurance of sterility throughout drug manufacturing is paramount to patient safety. Drug products must be free of contamination from microbes, particulates, and chemicals. To that end, we see continued and increased regulatory scrutiny, with adoption of US Food and Drug Administration (FDA) and European Medicines Agency (EMA) standards outside the United States and the European Union. To consistently make microbe-free products, drug manufacturers must implement robust, validated processes that include sterilizing filtration.
Sterilizing-grade filtration is used in a number of manufacturing process steps. In final fill operations, a sterile filtrate is necessary, so validation under worst-case conditions is required. For example, a high titer of a small organism may be used with the maximum allowed filtration time and maximum pressure. During intermediate operations, sterilizing-grade performance is highly desirable to keep a low microbial contamination risk and serve as insurance against potential in-process bioburden issues.
US FDA aseptic processing guidelines require a sterilizing filter to “reproducibly remove all viable microorganisms from the process stream, producing a sterile effluent” (1). For products that are not terminally sterilized, EMA guidelines state that solutions or liquids “can be filtered through a sterile filter of nominal pore size of 0.22μm or less, with [a filter] with at least equivalent microorganism retaining properties” (2). In brief, all microorganisms must be removed, and the microorganism retention properties of a filter must be well defined.
PRODUCT FOCUS: ALL BIOLOGICALS
PROCESS FOCUS: DOWNSTREAM PROCESSING
WHO SHOULD READ: QA/QC, PROCESS ENGINEERS, ANALYTICAL
KEYWORDS: FILTRATION, BUBBLE-POINT TESTING, VALIDATION
LEVEL: INTERMEDIATE
A technical report published in 1998 by the Parenteral Drug Association (PDA) and revised in 2008 described use and validation of sterilizing filtration as well (3). The most recent edition was developed in response to enhancements in filtration technologies and recent additional regulatory requirements for the pharmaceutical industry. The current version states that a sterile effluent must be validated in the intended application under worst-case conditions.
Failures in sterile filtration processes can be costly. Although the burden of meeting regulatory and industry requirements is on pharmaceutical manufacturers, filter suppliers must demonstrate that their products will help companies meet those stringent requirements. Here I describe the design and manufacture of filtration-system components to provide high assurance of sterility for aseptic processes by applying principles of quality by design (QbD) to the process.
Supplier ResponsibilityFilter suppliers must show that a sterilizing filter can remove all — not most — bacteria in a single process step and validate a sterile effluent in the intended application under worst-case processing conditions. Use caution when evaluating claims made for sterilizing-grade filters because a number of historical approaches do not meet the current minimum requirements. A nominal 0.2-μm filter size rating itself does not ensure sterile filtrate. The bacterial log reduction value (LRV) provides a good starting point, but it does not directly ensure that filtrate will be sterile. Another approach that is sometimes applied, serial filtration with larger pore sizes (e.g., two 0.45-μm filters in a series), may not deliver sterile effluent because no single step in the series ensures retention.

The minimum requirement is reflected in ASTM F838-05, which states that a filter must successfully retain all bacteria through the standard challenge test (4). Ideally, a filter will be validated with a defined safety margin above that minimum. A quantitative safety margin ensures low risk of failure. For bacterial retention, membrane bubble point is a key product property that can be used for establishing a safety margin.
Sterilizing filters occupy critical control points in a process. Beyond the minimum standard, it is important to know how consistent and reliable a sterilizing filter will be. Filters must be designed with a quantifiable high safety margin for bacterial retention and minimum loss of flow or processing time efficiency. Evidence of design conformance should be available from your filter manufacturer, and the risk of a filter being out of specification must be low.
Applying QbD to Filter DesignThe principles of the FDA’s QbD initiative can be applied to filter design and manufacture. In a QbD system, products are designed to meet patient requirements, and processes are designed to consistently meet product critical quality attributes. A manufacturer understands how components and process parameters affect product quality. Critical sources of process variability are identified and controlled. And the process is continually monitored and updated over time to ensure consistent quality.
Consistent with those principles, a “design space” (the combination and interaction of input variables and process parameters that have demonstrated quality assurance) is defined and validated (5). From that, a “control space” for on-going filter manufacturing is developed. For sterilizing filters, this is applied to three areas:
-
Membrane design and validation, during which membranes are developed with a quantified safety margin
-
Device design and validation, during which retention performance is verified
-
Manufacturing process control of critical process attributes (CPAs), during which continuous conformance to the design principles is monitored and ensured.
Designing a filter for sterility assurance begins with developing a manufacturing process within a well-understood design space. The process is carefully adjusted to make a series of membrane samples with different bubble points. Bubble point is a widely used, nondestructive integrity test that measures the largest membrane pore size to define size-based retention (3). For a given fluid and pore size, the pressure required to force an air bubble through the pores is inversely proportional to the size of those holes.
We then conduct ASTM bacterial retention testing on the membrane bubble-point series to measure organisms in the filtrate. A membrane is defined as “fully retentive” if it retains a challenge of >107 colony forming units (CFUs) per square centimeter of membrane area for an extremely small bacterium (Brevundimonas diminuta). For most applications, this represents a worst-case scenario. As Figure 1A shows, filtrate CFUs increase exponentially at decreasing bubble points. The log value of bacteria CFUs in the filtrate is used to set a filter specification to have high retention assurance under extreme ASTM conditions (Figure 1B). Sterility assuranceof >99.9% at 107 CFU/cm2 is desired. The safety margin comes from log(CFU) = –3. With that ma
rgin, zero CFU are expected 999 times out of 1,000 tests (Figure 1C). A more detailed explanation has been published elsewhere (6).
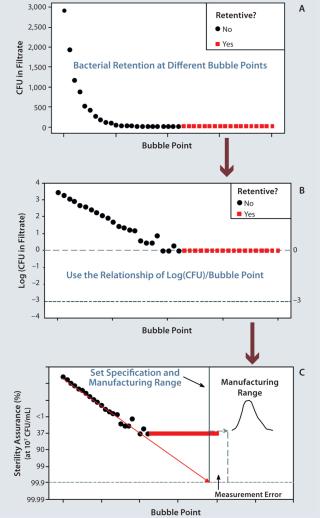
Once that safety margin has been established, we set a manufacturing range (the QbD control space). This range must be set well above the sterility assurance specification. It must be beyond the potential measurement error for bubble-point testing, and the manufacturing capability must be high.
Within those safety margins, filter membranes are routinely manufactured with >99.99% sterility assurance at a bacterial load of 107 CFU/cm2. Bubble point is controlled in process. All membrane rolls are tested, and their manufacturing process is adjusted in real time, if necessary. ASTM bacterial-retention performance is verified for each membrane lot.
Filter Device ManufacturingTo effectively support a sterile process, a membrane must be coupled with a complete device manufacturing process. For this process, QA-released membranes are 100% integrity tested. ASTM bacterial retention performance is verified on each lot of devices followed by a full panel of QA tests for endotoxins, extractables, flow rate, hydraulic stress, and resistance change after sterilization.
Final ValidationA filter device is then combined with user process conditions in a validation study to confirm that sterilizing performance is achieved for the intended application. Properties associated with the process for which a given filter will be used also must be examined during validation. They include physical properties such as temperature and time, bioburden profile and quantity, and any conditions that may adversely affect filter materials or filtration properties (e.g., radiation or extreme pH). As with filter design, the validation protocol uses a worst-case evaluation of both filter and process conditions.
Sterilizing filtration is a critical control point in biomanufacturing. A capable filter and thorough validation at worst-case filter and process conditions can provide sterility assurance at a high, known level with a low risk of failure.
Author Details
Mark Blanchard is a statistician in R&D design assurance at EMD Millipore, 80 Ashby Road, Bedford, MA 01730;