Fueled by a recent resurgence in public financing and compelling clinical data for indications as diverse as acute macular degeneration and pancreatic cancer, a growing number of cell therapies are driving toward pivotal clinical studies and commercialization. Although regulatory precedents have been set for various autologous and allogeneic products in the United States, Asia, and the European Union, regulatory guidance continues to evolve for a widening array of cell products.
Adult stem cells (e.g., mesenchymal stem cells), embryonic stem cells, induced pluripotent stem cells, genetically modified somatic cells, nongenetically modified cells, and cell–device combination products all require unique measures to ensure regulatory requirements for safety and efficacy. Beyond the inherent complexity of these emerging products, the migration of processes required to produce them paired with evolving technology platforms for manufacturing at scale offer additional challenges to their sponsors. Here we touch on some areas of concern for regulators and manufacturers in relation to the investigational-to-commercial transition.
Autologous or AllogeneicIn general, cell therapies are categorized as either autologous (patient specific) or allogeneic (universal or off-the-shelf) products. The regulatory pathway of an autologous product is determined by the degree of manipulation involved, the intended use of cells when returned to a patient, and association with a medical device if present. Cells that are minimally manipulated, intended for homologous use (performing their normal function), not associated with a device, and having no systemic effect upon return are considered to be “human cells, tissues and cellular tissue-based products (HCT/Ps),” as in Figure 1.
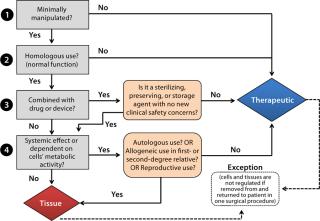
Sometimes referred to as “361 products” based on the section of the Public Health Service (PHS) Act from which the related legal authority is derived (1), 361 HCT/Ps are distinct from biological products or devices that require premarket approval. The predominant goal for their regulation is to prevent the introduction, transmission, or spread of communicable diseases. It requires establishment registration, defines donor eligibility standards, and requires that autologous HCT/Ps be manufactured in compliance with current good tissue practices (CGTPs) and additional requirements defined by FDA guidance documents (2). HCT/Ps regulated as drugs, devices, or biological products, and they also may have supplemental regulatory requirements such as good manufacturing practice (GMP) or quality system regulation (QSR). Except for HCT/Ps used in first- or second-degree relatives, allogeneic cell therapy products are generally regulated as biological therapeutics under applicable laws (the Food Drug and Cosmetic Act and Section 351 of the PHS Act) and regulations (21 CFR 1271, 600, and 200). If such a product is used in combination with a device or drug, then a determination must be made as to its classification and lead FDA review center (3).
Allogeneic and autologous products exhibit different features and represent unique challenges in the manufacturing of safe and efficacious products. Allogeneic products are considered to be less complex, often providing banks of cells that can be characterized for safety, and offer better prospects for scale-up/scale-out for the mass-produced products desirable for large indications. Issues for such “universal” products include potential immunogenicity, the possibility of tumor formation, and appropriate manufacturing platforms. Autologous product challenges include patient variability, manufacturing for large numbers of patients, and the need for a hybrid product-manufacturing service business model. Lesser concerns exist for immunogenicity or untoward side effects because of the patient-specific nature of such cells.
For both patient-specific and off-the-shelf products, US and international regulatory agencies share common concerns for cell therapies that must be addressed by their developers as companies navigate toward commercial use. Shared concerns include
-
appropriate collection and characterization of source cells
-
GMP facilities used for manufacturing
-
qualification and traceability of components, reagents, and raw materials
-
adequate characterization and testing of products for identity, purity, potency
-
stability of cells and cell lines over time
-
maintenance of desired differentiation properties or karyotypic alterations
-
assurance of product comparability through manufacturing changes.
Living cells are elegant products in that they are dynamic, can continue to grow and differentiate, and can even migrate within a patient’s body to a site of injury (Figure 2). Cells also can interact with their microenvironment. In some cases, a mixed population of cells may be responsible for a desired therapeutic effect. So cells represent complex products, which underlines the importance of understanding their sources and characteristics.

Source cells can present challenges to processing and logistics. Source variability (among patients) for autologous products and homogeneity of cell types must be addressed as much as possible to improve product consistency. Some manufacturers of patient-specific products have taken to providing both detailed training and well-designed collection kits to help address that source of variability. The relative scarcity of source cells and the availability of material at an appropriate stage of differentiation also can complicate process-development efforts and limit materials for use in validation of manufacturing processes.
For stem cell products, limited shelf life and limited numbers of cells can complicate quality control (QC) testing paradigms and stability determinations. For products that cannot be cryopreserved, a shipping range must be determined as well as transit times permissible for reaching patient treatment sites. Banked cells must be extensively characterized (according to prevailing regulatory guidance) for potential contamination with microbial or viral agents and for tumorigenicity, and tested in carefully designed safety studies using appropriate animal models. Studies documenting potential cell trafficking or distribution to nontarget sites are also required.
Source cells can present
challenges to product characterization. Determining critical quality attributes (CQAs) for complex products and developing assays for their potency are foremost concerns here. For example, Advanced Cell Technology (ACT) has developed a QC assay for guiding the process of expansion and harvest of retinal pigmented epithelial (RPE) cells derived from banked human embryonic stem cells (hESCs) used to treat ocular disease (4). The ACT assay relies on quantitation of melanin (which is naturally produced by the RPE cells) by absorbance at 475 nm. However, a simple assay that correlates to desired cell growth and differentiation may not always be available and is thus a benefit that manufacturers of other stem cell types may not enjoy.
Various design schemes have been applied to cell therapy manufacturing facilities, encompassing the strict environmental controls required for aseptic processing. Although relatively modest facilities may be adequate for early stage clinical programs, demands for later-stage manufacturing present hurdles to meeting regulatory expectations. Those include establishing appropriate facility and equipment performance standards, using closed systems wherever possible, implementing disposable equipment and process aids, and incorporating well-designed monitoring within facilities that are specifically designed for cellular aseptic processing. Some companies have established processing bays for manufacturing patient-specific products within a central manufacturing facility, whereas others have chosen to use individual modular suites dedicated to specific product-processing operations.
Contract manufacturers of cell therapies also must be prepared to address the differing facility requirements for autologous and allogeneic products that translate to varying suite size and equipment configuration. For example, WuXi AppTec has addressed such issues through a combination of multiple small and large suites, flexible expansion/processing areas, and high-yield cell-expansion platforms applying closed systems and disposable equipment throughout. Temporal and physical segregation of activities also allows for increased capacity within a manufacturing facility.
Components, Reagents, and Raw MaterialsWithout the safeguards that can exclude or inactivate potential contaminants for traditional pharmaceutical products, special attention must be paid to raw materials and components used in manufacturing cell therapies. These must be carefully evaluated and tracked as potential contributors of contaminants that could directly compromise product quality and safety. Enzymes, sera, matrices for cell attachment, cytokines and media components, culture vessels, tubing sets, bags, and other disposables must be carefully evaluated and tracked. Control measures required include generating bills of materials and establishing commodity specifications for safety, purity, and potency for all materials that document their specific sources, suppliers, and processing grades.
Vendor qualification programs must be established with product qualification programs that are routinely performed for incoming materials and components. Each new lot of material is subjected to testing or review to ensure that it meets specifications and is appropriate for use in manufacturing. Traceability (both forward and backward) is critical for all materials and components used in manufacturing (4). That challenge is met by some manufacturers through the use of material-processing databases or similar systems. Photo 2 shows a label generated by the validated material processing system at WuXi AppTec’s Philadelphia, PA, manufacturing facility. The barcode is used for scanning as materials advance from quarantine to released stores, and then for manufacturing.
Photo 1:

Testing bags, tubing, and culture vessels for extractables and leachables is required because such materials can potentially affect cell growth and viability. There is also some concern regarding the potential introduction of particulates from those materials to cell therapy products. Vendors of cell culture products are under increasing pressure to use “low-particulate” manufacturing methods for their products. Developers of cell therapies are expressing concerns that particles could interfere with biological efficacy of their products and possibly endanger patients.
Characterization and TestingSpecialized testing and QC paradigms are required for appropriately implementing in-process and product lot-release testing for cell therapies. An array of unique QC tests is used for stem-cell products: e.g., tests to screen for the absence of undifferentiated embryonic stem cells and tests to determine the extent of differentiation using gene expression, evaluation of cell morphology, protein production, and cell surface markers through fluorescence-activated cell sorting (FACS) analysis. Periodic testing for microbial contaminants is applied to cells or spent media for ensuring that aseptic conditions are maintained throughout processing.
Some cell products require release and administration to patients without completion of all required compendial testing. Rapid methods are frequently used, including assays that require extensive validation for use in lot-release testing of recombinant protein products. For example, the test method for mycoplasma detection based on polymerase chain reaction (PCR) has been used routinely in testing cell therapies after consultation with the regulatory agencies. Rapid microbial detection methods (e.g., the Bac-T Alert system from Biomerieux) also have been used in release testing of cell therapies, for which they enabled earlier reads on potential microbial contamination before patient administration while a 14-day sterility test (required by FDA regulation) continues to run.
Beyond actual test methods, consideration and compromise also must be made regarding sample volumes used in testing. In some cases, without test-volume adjustments the entire available cell product would be consumed. Cell products can be conserved through using surrogate samples (e.g., spent media from media exchange), testing feed media samples, and conducting multiple tests on a single sample.
Integration of specialized test methods within a manufacturing facility is of particular value for stem cell products with short shelf lives. Beyond the time savings in sample shipping or transfer, integrated test
ing operations permit necessary preplanning and coordination to reduce the time for product release to the shortest possible interval. Batch-record and documentation review by the quality organization requires seamless efficiency, as does the coordination of shipping logistics and communication with treatment sites. On-site, comprehensive testing capabilities adjacent to its cell therapy manufacturing facility has permitted WuXi AppTec to transport a fully released, formulated product to a clinical site and make it available to a physician within two hours of completing the final manufacturing step.
Many cell therapy products approaching late-stage studies or commercial use are targeting diseases with significantly large patient populations: e.g., diabetes; cardiac, neurodegenerative, and retinal diseases; and cancer. Processes that are adequate to supply clinical trials can become rapidly overwhelmed by commercial product demand. For products moving rapidly into advanced-phase clinical studies, the gap between clinical and commercial manufacturing process must be bridged quickly. In many cases, early stage processes using materials of inadequate quality for GMP compliance (e.g., research-grade components) or from inappropriate sources (e.g., bovine components from unacceptable sources of origin) can require extensive studies to document the comparability of the cell product from a “new” process.
Even for autologous products, scale-up or scale-out can be challenging. Process consistency across multiple manufacturing operators and process steps can be difficult, as can uniform adherence and compliance to established standards. Training of operators; quality assurance (QA) responsible for raw materials; and product, logistical, and support staff is critical to generation of consistently safe and effective cell therapies. Maintaining appropriate product segregation, preventing sample mix-ups, and ensuring clear communications and product distribution all become more difficult with expanding production. Systems for traceability of materials, reagents, and products must scale as well. Validated systems implemented at the earliest development stage provide a distinct advantage for forward integration of manufacturing processes. Modular and automated/robotic systems could provide solutions for some challenges to product consistency.
Allogeneic products are more amenable to automation and cost-of-goods benefits in scale-up, but they also present difficulties in transitioning from clinical to commercial scale. In some cases, migration to large scale can involve an entirely different platform for cell expansion, volume reduction, and harvest. Platforms such as single-use bioreactors with microcarriers may or may not yield the same product potency for adherent cells as flasks and trays. The difficulty here often lies in actually documenting comparability; tools and assays for assessing product quality are still in development. Lack of an extensive data set available for comparison creates a distinct disincentive to conduct such studies in early development. Given the relative novelty of commercial cell therapies, some companies wonder where regulatory agencies will draw the line between a process that produces a comparable product at larger scale and one deemed to be significantly different that it makes supplemental clinical studies necessary.
Less is often more. Innovative cell-expansion platforms such as Corning’s HYPER technology have been successfully used at WuXi AppTec to produce increased cell yields per square centimeter comparable to that produced in Thermo Scientific Nunc Cell Factory systems. We find that the Corning technology offers distinct advantages beyond providing greater surface area for cell growth within the same footprint. HYPERStack units are assembled by low-particulate methods, and the self-gassing attachement surface at each layer maintains a closed system without the need for active gassing. In our facility, we have successfully documented the utility of HYPERStack units as an alternative to our existing Cell Factory process for manufacturing a cell therapy product before initiation of phase 3 manufacturing.
Preliminary HYPERFlask studies ensured the appropriate growth and viability of therapeutic cells produced with this technology. Transition to the HYPERStack vessel was seamless. We verified cell growth, morphology, and viability in this scaled-up system as well as the cells’ unique product characteristics. New batch record documentation was revised, and manufacturing of phase 3 clinical material proceeded on the original production timeline. With the HYPERStack technology, suite productivity is significantly enhanced in our manufacturing facility without adding equipment or expanding processing areas. A single suite can accommodate 100 unit runs (100– 200 billion cells/100 HyperStack unit).
A wave of innovative cell therapies is presenting new technical and regulatory challenges, many of which are being effectively addressed. Regulatory agencies are evolving with the unique difficulties offered by these exciting therapies as they move from clinical to commercial manufacturing and are encouraging sound, risk-based evaluation of such products. Robust practices have been established that will permit licensure of safe and effective cell therapies. New scientific and technical developments are helping companies apply enhanced processes for manufacturing and QC of these potentially game-changing products.
Author Details
W. Alan Moore is vice president of strategic accounts and cell manufacturing, and John Bermel is director of manufacturing operations at WuXi AppTec, Inc.; 1-215-218-5500;