
WWW.GRAPHICSTOCK.COM
In part 1 of this two-part series, we outlined common challenges of technology transfer that are unique to the cell therapy industry and discussed strategies for success (1). Here, we delve even further into best practices and highlight key strategies for technology transfer that should be considered along the path to success. Creating a strong foundation for technology transfer will streamline clinical manufacturing processes and help position therapeutic products for long-term success. Below are key criteria for success.
Confirm Transfer Acceptance Criteria
Creation of a program charter is a perfect place to start discussions about acceptance criteria for activities to be performed during the course of a project. Acceptance criteria help you ascertain whether an ongoing technology transfer is successful at a given time. An absence of clear acceptance criteria before an activity leads to misaligned expectations of success, resulting in frustration and disappointment on all sides.
Acceptance criteria should be based on known data (usually generated during product development at a transferring and/or receiving site) and aligned to the project stage. Time should be invested as early in the program as possible to define acceptance criteria and how they will be documented. That includes formal training and development reports used to document a transfer process as well as the process description and associated documentation.
Process Development and/or Improvements
Status and robustness of transferred processes can vary extremely, from client to another and often correlate to clients’ understanding of good manufacturing practices (GMPs) and what is appropriate for each phase of a planned clinical trial. Processes that have been adapted to work well in R&D and academic/hospital GMP facilities are not always directly transferable to a GMP manufacturing setting. Anticipated and unanticipated adaptations might be involved. Some examples follow.
Changes in Incoming Product Materials: Incoming product materials collected from healthy individuals might differ from those collected from patients undergoing treatment. This carries a risk of unanticipated effects on manufacturing processes and analytical methods.
Changes in Scale: For patient-specific products, subbatch processing (splitting a lot to compare processes at transfer and receiving sites) might not be feasible. For allogeneic products, relatively small changes in scale can have unanticipated effects on a process.
Changes in Procedure: Changing a bioreactor or expansion vessel type, for example, or transitioning from tissue culture flasks to culture bags is highly likely to affect a process.
Changes in Raw Materials: A conversion from research-grade to CGMP-compliant in-process materials can have unanticipated effects on a process.
Changes in Timing Before and After Processing: As mentioned in part 1, for example, a change in proximity of raw material collection and manufacturing site can have a significant impact.
Manufacturing timing can change in a cleanroom environment because of CGMP constraints. For example, movement in and out of a biosafety cabinet (BSC), extra time needed for documentation, and a need for real- time process verification by an independent operator all can add time. In-process monitoring of process parameters (e.g., cell counts) normally performed by an operator in an R&D laboratory often takes longer when samples must be sent out of a controlled-environment room (CER) to a quality control (QC) associate in another laboratory. So hold steps relying on a cell count can be significantly increased.
Changes in Product Storage and Shipment: Cell therapy products often are developed to be manufactured and shipped at a controlled temperature to a clinical site. But they have a limited shelf life as a result, which creates supply chain risks and potential product failure. Shipping and stability (including cryopreservation) studies typically are used in Stage 4 (Figure 1) to direct implementation of beneficial changes.
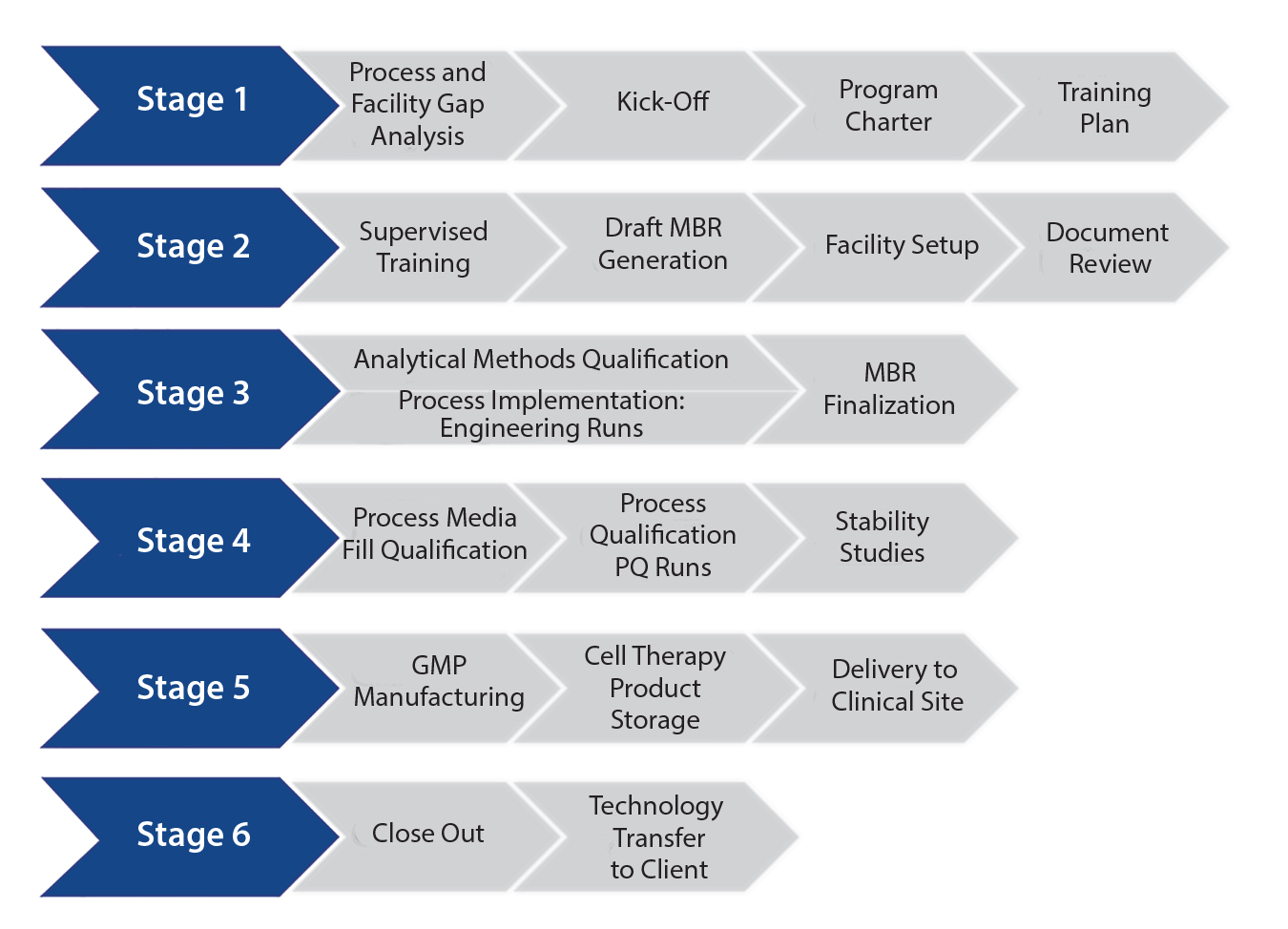
Figure 1: Streamlining technology transfer
Changes in Analytical Assays: Development of more robust analytical methods (typically involving new equipment and protocols) can be evaluated and integrated into a technology transfer strategy.
Development requires process change, although such changes are best avoided during technology transfer. Process changes can seriously affect a program, and they are ideally implemented once transfer is complete under change control. A sponsor should understand that a contract development and manufacturing organization’s (CDMO’s) facility and operations do not need to be an exact replica of its own. Instead, the collective team should ensure that
- a process being transferred is robust and well-understood, with data-driven acceptance criteria
- the CDMO’s facility, material flow, and environmental controls meet set process requirements
- equipment meets process-specific specifications
- QC testing and analytical procedures meet established product and assay parameters
- the CDMO’s quality systems provide the relevant controls needed for implementation
- change control and quality by design (QbD) principles are stringently applied to unit operation (UO) inputs and outputs to demonstrate comparability through reliable assays.
Training
The training process should be documented in a training plan with predefined acceptance criteria used to measure success. It should be compiled by collaboration among the sponsor, transferring site (if different), and CDMO. For example, before being assigned to a given program, PCT team members will have undergone comprehensive training that includes but is not limited to an understanding of GMP requirements, cleanroom operations, use of common equipment, and aseptic techniques (assessed using media fill qualification). QC associates have complementary training in common QC assays and associated equipment.
At the start of a program, paper training (reading client-supplied materials) should provide the first exposure to the new process, and training should continue to develop throughout the program. Hands-on training performed by transferring-site staff allows a CDMO to capture practices, experience, and knowledge not included or difficult to define in standard operating procedures (SOPs). It is also an opportunity for a CDMO to audit the process before transfer. Setting prospective acceptance criteria and expectations will help guide and measure training success.
Follow-up training at a CDMO’s facility allows a sponsor (or transferring site) to observe CDMO staff and provide additional technical feedback that will be essential for successful process implementation. Such training also allows a sponsor to develop trust and confidence in a CDMO’s technical proficiency. The CDMO staff will continue to develop their knowledge further as SOPs, work instructions (WIs), and master batch records (MBRs) are compiled and processes implemented. The outcome of all training activities should be documented in a training report and the success of those activities evaluated and agreed upon before moving to process implementation.
Process Implementation
A sponsor and/or transferring site and the CDMO receiving site together should detail the process implementation method in an engineering run protocol, with predefined acceptance criteria used to measure success. During this stage, analytical methods normally undergo qualification using samples generated during engineering runs as test articles.
The combined team should perform a phase-appropriate assessment of the qualification/ validation level for the cleanroom, equipment, ancillary and manufacturing procedures, and analytical methods suitable for implementation before beginning engineering runs. That assessment should account for regulatory requirements while focusing on practical concerns in the context of the process being transferred (risk assessment). A CDMO’s prior experience will be particularly valuable at this stage.
Performing an additional engineering run provides additional risk mitigation. During that run, a transferred process is performed in the cleanroom environment in Stage 3 before proceeding to PQ. The practice of executing a “dress rehearsal” also provides an opportunity to continue comparison with sponsor- and/or transferring site-manufactured product and process. Final adjustments to the process can be made at this point, along with adjustment of relevant documents and retraining deemed necessary.
During process implementation, acceptance criteria are normally narrower than those used during training runs and are again used to measure success. Data and observations from engineering runs should be captured in a report that will be jointly reviewed by both parties before making a decision to move on to PQ runs. Failure to make that assessment and jointly agree on a decision to move forward — based on timeline restrictions or other external factors — can result in failures down the line that are difficult to overcome.
Process Qualification
A sponsor and/or transferring site and CDMO receiving site together should detail the PQ processes in a PQ run protocol, with predefined acceptance criteria used to measure success. PQ runs are performed according to a prospectively determined set of criteria following an MBR. By contrast with engineering runs, PQ runs are “locked down” following process implementation. PQ runs follow the finalized process that will be used for clinical manufacturing and should be treated as such. These runs provide an opportunity to perform all finalized steps in sequence, from beginning to end (including QC testing). Whenever possible, they should simulate the clinical process from raw material collection and shipment through to final product release and shipment.
Just as with engineering runs, data and observations from PQ runs should be reviewed by both parties collaboratively before making a joint decision to move on to clinical manufacturing. Clients typically use information in PQ reports to support investigational new drug (IND) applications (or equivalent) and comparability assessments based on prospectively set criteria.
Once PQ runs have been performed, a media-fill qualification of the process based on phase-appropriate risk assessment might be necessary. In such cases, the process (or parts of it) is replicated using tryptic soy broth (TSB) in lieu of medium and buffers used during clinical manufacturing. Samples are collected at regular intervals throughout this process and tested for sterility.
Clinical Manufacturing
A successful, streamlined program ultimately reaches GMP manufacturing (Stage 5, Figure 1) cost-effectively. It should consist of a firmly established, robust, and reliable qualified process with analytical assays, governed by an MBR and other controlled documents to allow production of tens to hundreds of GMP cell therapy batches at a CDMO site.
During implementation of a qualified process for the first few patients, a need often arises for ongoing troubleshooting and optimizing. That is based on the use of clinical sites for collection and dosing as well as the use of clinical material for the manufacturing process itself. In such cases, critical process parameters (CPPs) and unit operations can provide an ongoing and disciplined approach to maintaining process control.
As previously noted, before clinical manufacturing, technology transfer often is performed using raw material collected from healthy individuals by nonclinical collection facilities. However, materials from affected patients (clinical trial subjects) can vary considerably from those collected from healthy individuals. Such differences can result from the disease state of patients or from collection procedures implemented by clinical facilities. Either way, the result is a raw material that is different from what the CDMO had previously encountered during technology transfer. Such differences can affect the manufacturing process or the ability of a QC laboratory to reliably perform its assays.
Ongoing discussion and dialog among all parties is critical at this time. Tackling problems (if and when they arise) requires a calm and methodological approach. Once those issues have been ironed out, clinical manufacturing usually becomes routine.
However, a transferring site should nonetheless ensure that a process doesn’t become “trapped” at the CDMO, developing barriers to the “transfer-out” of the process to another sponsor-designated facility. Stage 6 of Figure 1 incorporates a defined and structured strategy for successful process transfer out of a CMDO if and when required.
Risk Management
Gaps and risks should be evaluated regularly and consistently at the beginning of technology transfer and move forward consistently. Large and expected risks are proactively addressed usually at the start of technology transfer. Small or unforeseen risks that are not identified early can have unanticipated negative effects on a project and could even stop the project altogether. For example, single-vendor suppliers of common and critical materials are a well-known risk. A lot failure by a vendor or a company acquisition can result in a raw material becoming no longer available, and an alternative will need to be found or the process changed or adapted. That could lead to the need for comparability studies before continuing with patient accrual in a trial, resulting in unanticipated delays. The “Technology Transfer Case Studies” box describes other potential risks and mitigation strategies.
Strategy for Success Cell-based therapeutics come with inherent variability and present unique challenges for technology transfer processes. Having a structured, strategic approach will minimize risk and enable transferring sites to generate greater value for process and product. A clear, long-term vision of the process and product — combined with CDMO expertise to navigate the technology transfer pathway — will help position a cell-based therapeutic for commercial success.
| Technology Transfer Case StudiesCritical Raw-Material Shortages Example: Custom order for medium or supplement Assumption: In-house stocks or secured lots will be sufficient should a manufacturer have a lot failure. Risk of Failure: Medium probability, high impact Mitigation: Secure lots with supplier Mitigation Breakdown: Supplier had multiple successive lot failures. PCT–Recommended Mitigation: Identify backup independent suppliers for critical materials.Noncritical Raw-Material Shortage Example: Normal saline Assumption: A commonly used raw material will never be in short supply; multiple suppliers can be used should one supplier run out of material. Risk of Failure: Low probability, high impact Mitigation: Multiple suppliers manufacture and sell normal saline. Mitigation Breakdown: A major US supplier reduced production that resulted in a severe shortage PCT-Recommended Mitigation: Monitor supply chain of all materials carefully and increase in-house supplies and/or identify backup suppliers for noncritical raw materials.Importance of Tribal Knowledge Example: Trypan blue exclusion assay for the determination of viable-cell concentration and percent viability Assumption: Standard procedures are performed in the same way in different laboratories. Risk of Failure: Low probability, high impact Mitigation: Staff at receiving site are highly trained in this assay and shown to have similar competency to each other by comparing similarity of cell counts and viability on the same samples. Mitigation Breakdown: Transferring site had in-house modifications to the assay not transferred to the receiving site PCT-Recommended Mitigations: Share data (ranges and variability) generated by a transferring site, and use this to assess data generated by the receiving site. Train receiving site staff at the transferring site facility.Logistics and Product Stability Example: Shipment of incoming raw material (e.g., apheresis) to manufacturing site or final product to clinical site Assumption: Shipment will always be completed within 24 hours because shipping companies state that they can perform overnight shipment and delivery; 24-hour stability studies and shipping qualifications performed Risk of Failure: Low probability, high impact Mitigation: Careful monitoring of shipment in collaboration with shipping company will ensure delivery within a 24-hour window. Mitigation Breakdown: During extreme weather events, shipments can be delayed while weather conditions improve. Shipping companies can lose track of shipments. Shipments can be held up in unexpected locations. PCT-Recommended Mitigations: Do not underestimate the importance of good logistics management of shipments. Use a courier service (rather than a shipping company) to transport critical raw material and/or final product. Design and qualify shipping procedures for as long as possible (not <48 hours). |
Reference
1 McIntyre C, Sumen C. Are You Ready for a Tech Transfer? Part 1: Challenges and Critical Factors for Success in Cell-Therapy Development. BioProcess Int. 13(4) 2015: S44–S47.
Corresponding author Catherine McIntyre, PhD, is director of technical operations at PCT West, 291 N. Bernardo Avenue, Mountain View, CA 94043. Cenk Sumen, PhD, is manager of technology and business development at PCT East, 4 Pearl Court Suite C, Allendale, NJ 07401.