WWW.GRAPHICSTOCK.COM
The development pathway of a biosimilar is unlike that of a novel biotherapeutic. Many regulatory authorities reference a stepwise approach to establishing biosimilarity. Analytical requirements are greatly increased before a product enters clinical testing. Enhanced analytical efforts entail physical, chemical, and biological characterization of a biosimilar product compared with an originator reference product. Strategies at this early stage must include intensive characterization of multiple batches to determine variability of quality attributes.
Here I address some issues involved in meeting regulatory characterization expectations by answering the following questions: What regulatory guidelines are associated with structural characterization and comparability/biosimilarity testing? What complicates the characterization of complex (glyco)protein products? When is analytical characterization required? And which techniques (old and new) are suitable for this application?
Evolution of Regulatory Requirements
The European Union established the first biosimilar guidelines for “similar biological medicinal products” (biosimilars) about a decade ago. The European Medicines Agency (EMA) pioneered a novel approach to authorization, which initially requires physical, chemical, and biological characterization of a biosimilar in direct comparison with a reference product. Following that extensive characterization, nonclinical and clinical data should demonstrate the same safety and efficacy profiles as for the originator. However, the premise of this approach is that the amount of nonclinical and clinical data required will be much less for a biosimilar than for an innovator molecule; hence, the time scale should be reduced.
Effective in 2005, the initial “overarching” principle-defining guideline (CHMP/437/04) was followed by further general guidelines on quality, nonclinical, and clinical issues. Subsequently, data requirements specific to product classes were published for a number of molecules: human growth hormone (HGH), granulocyte colony-stimulating factor (G-CSF), epoeitin (EPO), follicle-stimulating hormone (FSH), insulin, interferons alpha and beta, low–molecular-weight heparins, and monoclonal antibodies (MAbs).
Many of those original regulatory guidelines (both “overarching” and product-specific) have since been revised. The most recent of these is the nonclinical/clinical guideline that became effective in July 2015. You can find all the EMA guidelines with current revision concept papers online (1). These updated documents reflect learnings gained from over 10 years of EMA experience in reviewing biosimilar marketing applications in Europe.
For example, the revised quality guideline (EMA/CHMP/ BWP/247713/2012 Revision 1, effective 1 December 2014) now contains additional specific details relating to the comparability exercise, such as the following:
- State-of-art analytical, orthogonal methods must be used; it is not acceptable to use only methods that were in place when the originator product was authorized.
- Comparative characterization studies should include assessment of composition, physical properties, primary and higher-order structures, purity, product-related substances (e.g., isoforms), impurities, and biological activity with “sufficiently sensitive analytical tools.”
- Quantitative ranges should be established for quality attributes.
In the United States, the Biologics Price Competition and Innovation Act (BPCIA), part of the Patient Protection and Affordable Care Act, was signed into law by President Barack Obama in 2010. It introduced an abbreviated approval pathway for biosimilars: the 351(k) route of the Public Health Service (PHS) Act. As in Europe, this pathway requires comparing a biosimilar molecule with an originator reference product that was approved under the normal 351(a) route. Biosimilar applications must include information demonstrating
based on data derived from: analytical studies that demonstrate that the biological product is highly similar to the reference product notwithstanding minor differences in clinically inactive components, animal studies (including the assessment of toxicity); and a clinical study or studies.
Note that the US Food and Drug Administration (FDA) may determine at its own discretion and case-by-case whether any requirements are unnecessary.
One unique aspect of the US legislation is its provision for two levels of approval: “biosimilar” and “interchangeable biosimilar.” Interchangeable products may be substituted for a reference product without intervention of a prescribing healthcare provider. For such products, additional data would be required (possibly including clinical “switching studies”).
The first FDA guidances to help manufacturers develop biosimilars were drafted in 2012 and covered scientific and quality considerations along with common questions and answers. Their final forms were issued earlier this year. Further guidances have been released to discuss scientific meetings (2013) and clinical pharmacology data demonstrating biosimilarity (2014). An additional draft Q&A document was issued in May 2015. All of these can be found online (2).
Those documents introduced several important concepts concerning the scientific rationale for demonstrating biosimilarity. The FDA will consider the “totality of data and information submitted” in assessment of biosimilar applications and will allow reference to publicly available information regarding previous determinations that a reference product is safe, pure, and potent. The agency also suggests that companies use a “meaningful fingerprint-like analysis algorithm” to design and help reviewers interpret an analytical data package. That recommendation is echoed in an introduced concept: Four possible outcomes follow assessment of comparative analytical characterization data — not similar, similar, highly similar, and highly similar “with fingerprint-like similarity.” That determination will dictate the level of information required in further steps toward approval.
Why Is Characterization Such a Challenge?
Unlike chemically synthesized small-molecule drugs, biological products are “manufactured” by living systems such as genetically engineered bacteria, yeast, fungi, or mammalian cells. During or after transcription of DNA code through messenger RNA into an amino-acid sequence, intracellular processing events occur to confer biological activity upon many final proteins. Cotranslational and posttranslational modifications (PTMs) of a molecule change its primary structure, and because the changes are not predictable from a gene sequence, expressed protein products must be assessed. The “Protein Modifications” box lists common protein modifications.
Glycosylation is arguably the most important PTM. Attachment of carbohydrates to a protein chain certainly increases its complexity and adds additional layers of potential product heterogeneity. Glycoproteins come in mixtures of glycoforms: the same polypeptide with different glycan units attached. The amounts and distribution of sugars on a protein backbone depends on the cell type in which it is expressed and the cells’ physiological state, making it easily affected by manufacturing/process conditions.
Glycosylation is important for many reasons. Its presence is required for biological activity in some molecules (e.g., EPO) and thus affects therapeutic efficacy. However, certain nonhuman glycan structures can be immunogenic in humans, so it is essential for biopharmaceutical manufacturers to screen glycoprotein products for them.
When Is Analytical Characterization Required?
As outlined above, the development pathway of a biosimilar is specialized, with much greater requirements for physicochemical and biological analytics at early stages than for novel biotherapeutics. Figure 1 illustrates a general stepwise process as referenced by many regulatory authorities.
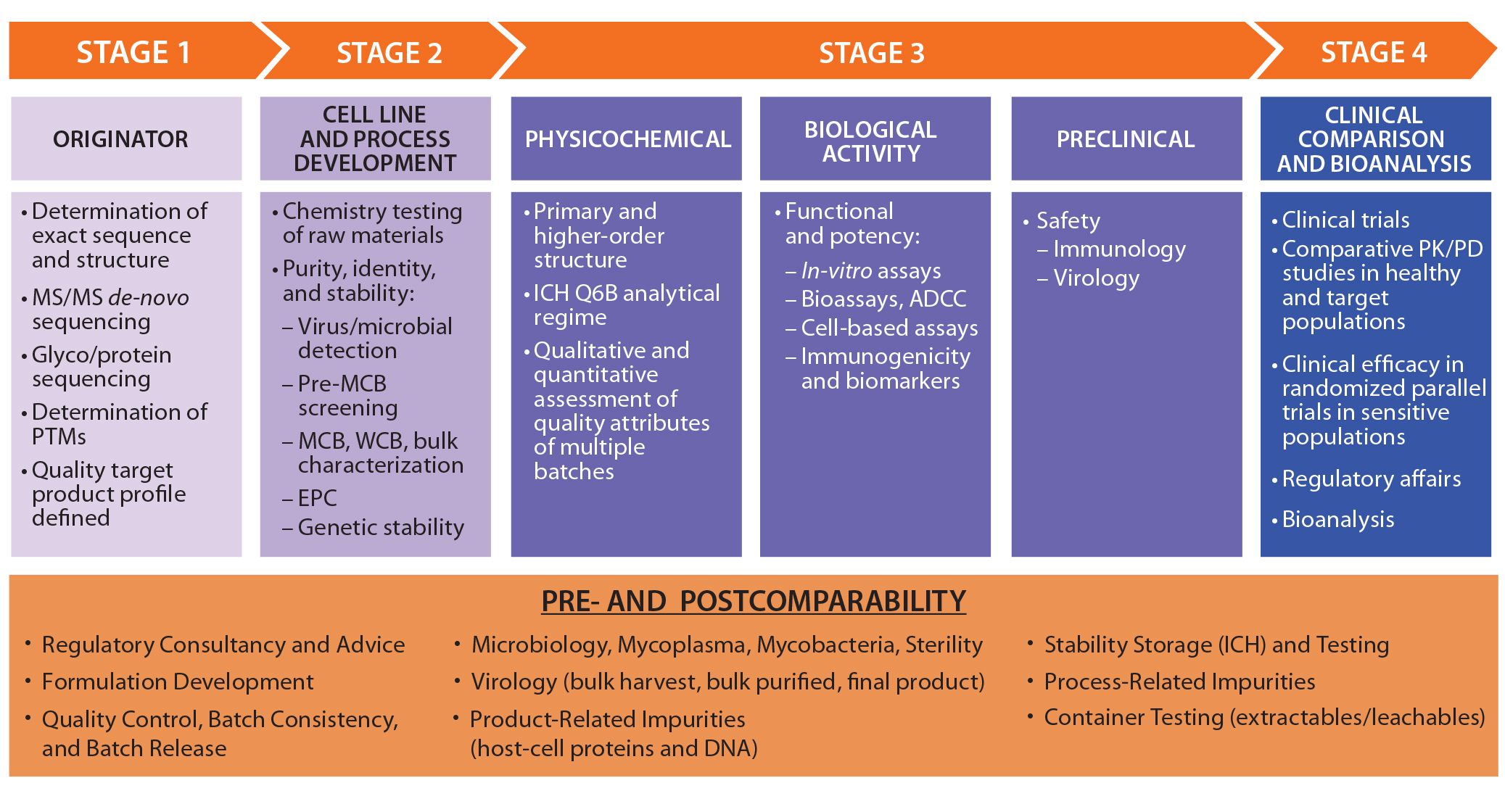
Figure 1: Side-by-side comparison; ADCC = antibody-dependent cell-mediated cytotoxicity, EPC = end-of-production cells, MCB = master cell bank, PK/PD = pharmacokinetcs/pharmacodynamics, PTMs = posttranslational modifications, WCB = working cell bank
Characterization efforts begin before a biosimilar is manufactured. Initially, the target originator (reference) molecule must be intensively studied to determine the variability of its quality attributes. Multiple originator batches are studied to determine the amino-acid sequence and related PTMs. Determining the exact amino-acid sequence entails de novo sequencing approaches involving tandem mass spectrometry (MS/MS). It is a crucial step because the resulting data form the basis of a quality target protein profile (QTPP) for a biosimilar. At the cell-line and process development stages of biosimilar manufacturing, characterization surveys can help in the selection of an appropriate cell line to design biosimilarity into a product from the outset.
Once a biosimilar product is expressed, regulatory guidelines require comparative side-by-side analysis with an originator molecule for quality, safety, and efficacy. This is in addition to the usual complete chemistry, manufacturing and controls (CMC) data package for a biologic molecule. It raises questions about the choice of reference material: From which regulatory jurisdiction should it be derived? How many batches of biosimilar and originator products should be compared? And what changes in the originator might have occurred over the lifetime of that product? The objective is to determine whether the proteins have the same biochemical, biophysical, and physiological attributes.
There is also a requirement to identify product and process-related variants and impurities. FDA guidance emphasizes a stepwise approach to study design, which should allow for evaluation of residual uncertainty at each step. If differences are observed, then their potential impacts on safety, purity, and potency must be fully addressed through further experimentation.
Biosimilarity assessment requires strategies for both primary and higher-order structure, which can be determined using physicochemical analytical techniques with appropriate sensitivity. It can be a challenge to decide which techniques to use, bearing in mind the “fingerprinting” concept. Specification guidelines from the International Conference on Harmonisation of Technical Requirements for the Registration of Pharmaceuticals for Human Use (ICH) cover physicochemical characterization, with a useful appendix for ensuring that all of a molecule’s physical attributes are covered (see “ICH Q6B” box).
| ICH Q6B Test Methods |
| ICH topic Q6B specifications — test procedures and acceptance criteria for biotechnological/biological products (3): |
Structural characterization and confirmation
|
Physicochemical properties
|
| Protein Modifications |
| These are some common cotranslational and posttranslational cellular protein modifications: Acetylation, Acylation, Amidation, Carbamylation, Carboxylation, Deamidation, Formylation, Glycation, Glycosylation, Methylation, Methionine, Oxidation, Norleucine, Phosphorylation, and Sulphation |
Comparative biological functional and binding assays are conducted in parallel with the quality structural comparison. Only once biosimilarity has been established will regulatory authorities allow progression to clinical evaluation in a product’s intended population. Pharmacokinetic and pharmacodynamic phase 1 trials are followed by clinical efficacy and safety studies in phase 3, as required.
Which Techniques Are Suitable?
Analytical methods — both classical and emerging technologies — can be applied to comparative analysis of glycoprotein biosimilars. A thorough understanding of both the capabilities and limitations of each technique is critical to determining the most appropriate strategies. A comparability study should be comprehensive enough to minimize the possibility of undetected differences. The “Analytical Tools Box” (below) lists a selection of potential “state-of-the-art” analytical methods. Many techniques in that inexhaustive list are based on mass spectrometry (MS). Over the past 30 years, such methods have become essential in (glyco)protein structural determination.
| Analytical Tools |
| Amino Acid Sequence and Modifications: mass spectrometry (MS), peptide mapping, gas-phase sequencing (GPS), and chromatography |
| Glycosylation: anion-exchange chromatography, enzymatic digestion, peptide mapping, capillary electrophoresis, and MS |
| Folding: MS S–S bridge determination, calorimetry, hydrogen–deuterium exchange (HDX) and ion-mobility MS, nuclear magnetic resonance (NMR), circular dichroism, Fourier-transform spectroscopy, and fluorescence |
| PEGylation and Isomers: peptide mapping and chromatography |
| Aggregation: analytical ultracentrifugation, size-exclusion chromatography with multiangle light scattering (SEC-MALS), field-flow fractionation (FFF), dynamic light scattering (DLS), microscopy, and transmission electron microscopy (TEM) |
| Proteolysis: electrophoresis, MS, and chromatography |
| Impurities: proteomics, immunoassays, and metal and solvents analysis |
| Subunit interactions: chromatography, ion-mobility MS |
| Heterogeneity of size, charge, and hydrophobicity: chromatography, gel and capillary electrophoresis, light scattering, ion-mobility MS, capillary electrophoresis and electrospray ionization with MS |
Comparative analysis of a MAb biosimilar can be even more challenging. The complexity and size of MAb molecules can be extensive, as can their potential structural variations in quality attributes (e.g., deamidation, glycosylation, C-terminal clipping, and so on), leading to vast numbers of potential variations to be deduced.
Guided by ICH Q6B (3), scientists can measure intact molecular-mass measurement on the whole molecule (or on reduced and released light and heavy chains) as a starting point. Using modern mass spectrometers, they can obtain well-resolved and accurate data at 150 kDa, allowing assessment of glycoforms. This intact mass is a very useful starting point as a comparison tool, allowing companies to study several batches of antibodies.
That analysis is usually followed by N- and C-terminal sequence determination and peptide-mass mapping. The latter is a particularly powerful routine structural confirmation tool. In essence, a protein is digested after reduction and alkylation of disulphide bridges, using specific proteases to produce a mixture of peptides that can be analyzed with MS to provide a mass fingerprint or map. Molecular change would alter the mass map, making it a very effective identity test. In practice, mapping large molecules such as MAbs requires several proteolytic digests to be performed in parallel. The resulting peptides also can be separated with online liquid chromatography (LC) with MS. With high-energy MS/MS sequencing, we can confirm the amino acids in the protein sequence. A variation of the peptide mapping strategy with the initial map conducted under nonreducing conditions can be used to locate and assign disulphide bridged peptides and free thiols.
Determination of a glycoprotein’s glycan moiety is as important as protein sequencing. The carbohydrate profile of a biosimilar may not necessarily have to be the same as that of the originator protein; however, further studies would then need to prove that the different glycoforms affect neither safety nor efficacy. Glycosylation influences the pharmacokinetics of certain proteins, so comparative PK/PD studies should be performed to establish whether they are within an acceptable range.
For glycoproteins, the following should be determined at minimum: carbohydrate content (neutral sugars, amino sugars, and sialic acids); structure of the carbohydrate chains, oligosaccharide pattern, antennary profile, and linkages; and glycosylation sites on the protein chain. A number of glycomic and glycoproteomic strategies can be used to interrogate the glycan moiety. Starting at the intact glycoprotein level, screening for the extent and heterogeneity of glycosylation can be performed using matrix-assisted laser-desorption ionization (MALDI) and/or electrospray MS both before and after deglycosylation. That would be followed by quantitative monosaccharide composition and sialic acid content analysis with gas chromatography and mass spectrometry (GC/MS) and high-performance anion-exchange chromatography with pulsed amperometric detection (HPAEC-PAD).
Further digestion and mass-mapping LC–MS strategies provide qualitative, site-specific glycosylation information for both N-linked and O-linked structures. N-linked glycans can be removed enzymatically, O-linked glycans with reductive beta-elimination from the digested glycoprotein, then analyzed using MS with, for example, ion-exchange chromatographic methods on both underivatized and derivatized samples. Doing so will provide information about glycan profiles: quantitation, composition and sequence, antennary profiles, intermonosaccharide linkages, and glycosylation sites. It is particularly important to assess the stereospecificity of oligosaccharide antennae linkages because certain glycotopes (e.g., Gal-alpha-1,3-Gal) can promote antigenic stimulation in humans.
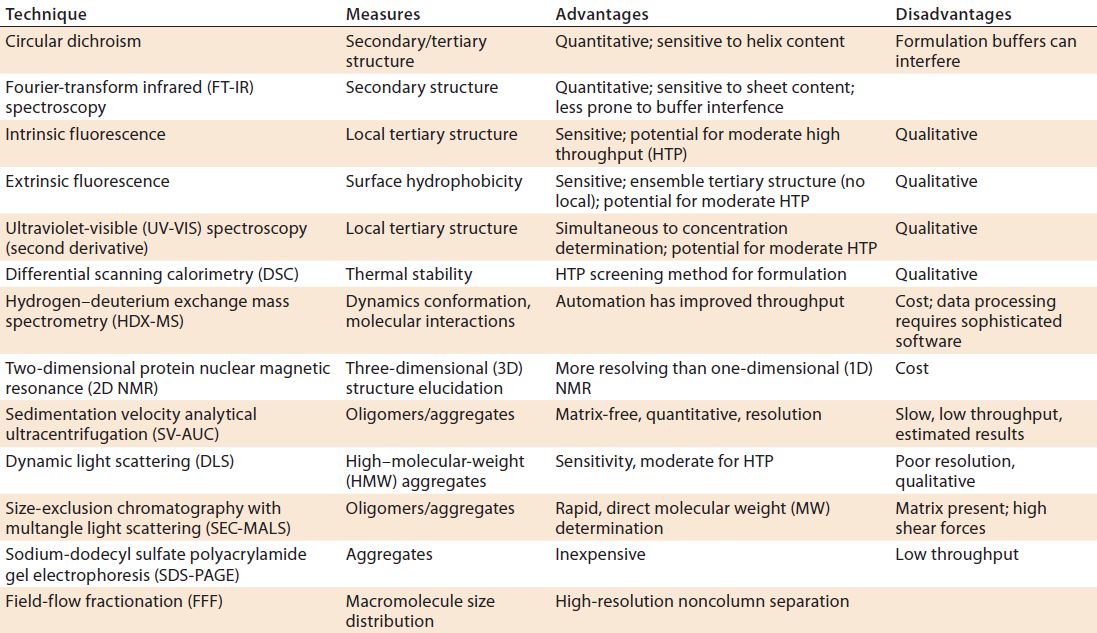
Table 1: Biophysical techniques for higher-order structure, conformation, and aggregation
Finally, three-dimensional (3D) structure determination (secondary, tertiary, and quaternary) requires orthogonal biophysical tests such as those listed in Table 1. In addition, techniques such as analytical ultracentrifugation, dynamic light scattering (DLS), and fluorescence resonance energy-transfer methods measure (undesirable) aggregates that may be formed. It is important to remember that that no single technique will provide full unequivocal information and that each has its pros and cons. Regulators are aware of those limitations and expect a careful selection of orthogonal methods. For example, routine size-exclusion chromatography (SEC) should be complemented by a noncolumn technique such as sedimentation-velocity analytical ultracentrifugation (SV-AUC) or field-flow fractionation (FFF).
Linking higher-order structure with biological activity is increasing in importance. Several techniques are emerging from research applications to probe the structure of proteins. These include hydrogen–deuterium exchange mass spectrometry (HDX-MS) and two-dimensional (2D) nuclear magnetic resonance (NMR) spectroscopy to provide greater resolution of complex structures.
Emphasis on Complementary Methods
Extensive structural and functional analytical characterization data form the foundation of a biosimilarity evaluation. Multiple orthogonal analytical methods should be used to compare biosimilar and originator molecules. Strategies for primary and higher-order structure determination are required to define a product “fingerprint” using methods that are sensitive enough not only to demonstrate similarity, but also to detect small changes in posttranslational modifications such as glycosylation.
A comprehensive characterization package provides biomanufacturers with an understanding of the relationships between identified critical quality attributes (CQAs) and clinical safety and efficacy profiles on which both biosimilar manufacturers and regulatory authorities can act.
References
1 Multidisciplinary: Biosimilars. European Medicines Agency: London, UK, www.ema. europa.eu/ema/index.jsp?curl=pages/regulation/ general/general_content_000408. jsp&mid=WC0b01ac058002958c.
2 Biosimilars. US Food and Drug Administration: Rockville, MD, www.fda.gov/Drugs/GuidanceCompliance RegulatoryInformation/Guidances/ ucm290967.htm.
3 ICH Q6B. Specifications: Test Procedures and Acceptance Criteria for Biotechnological/Biological Products. US Fed. Reg. 18 August 1999: 44928; www.ich.org/ fileadmin/Public_Web_Site/ICH_Products/ Guidelines/Quality/Q6B/Step4/Q6B_ Guideline.pdf.
Fiona M. Greer, PhD, is global director of biopharma services development for SGS Life Sciences, 1 Place des Alpes, PO Box 2152, 1211 Geneva 1 Switzerland; 41-22-739-9111; fax 41-22-739-9886; www.sgs.com. This article is based on a presentation at the March 2015 Biotherapeutics Analytical Summit in Baltimore, MD.